
10 Dec 2025
Accurate prediction of the frequency response of quantum dots under electromagnetic radiation is essential for investigating absorption spectra, excitonic effects, and nonlinear optical behavior in quantum dots and semiconductor nanoparticles. The polarization propagator provides a rigorous framework for evaluating these properties, but its construction is computationally demanding. Challenges arise from the level of electron correlation, the size of the excitonic basis, and the cost of evaluating two-electron integrals. This work addresses these difficulties by developing first- and second-order frequency-dependent polarization propagator calculations for PbS and CdS quantum dots. The propagator is formulated using the electron propagator approach and expressed as the resolvent of the Hamiltonian superoperator. Light-matter interaction is treated using the dipole approximation and represented in a particle-hole excitation operator basis. The correlated ground state is treated at the MP2 level, and all response-matrix terms up to second order in the fluctuating potential are included. A frequency-dependent inverse Krylov subspace method is derived and combined with the folded-spectrum technique to isolate excitation energies within a chosen frequency window. This strategy avoids full diagonalization of the response matrix and significantly reduces computational cost for large systems. The method is implemented in a matrix-free manner in which no explicit response matrix is assembled, and all operations rely on matrix-vector products. UV-VIS excitation spectra of PbS and CdS quantum dots were computed, demonstrating that the inverse Krylov subspace projection approach provides an efficient and accurate approximation for excitation spectra when full diagonalization is computationally prohibitive.

28 Jun 2022


A large body of knowledge about magnetism is attained from models of
interacting spins, which usually reside on magnetic ions. Proposals beyond the
ionic picture are uncommon and seldom verified by direct observations in
conjunction with microscopic theory. Here, using inelastic neutron scattering
to study the itinerant near-ferromagnet MnSi, we find that the system's
fundamental magnetic units are interconnected, extended molecular orbitals
consisting of three Mn atoms each, rather than individual Mn atoms. This result
is further corroborated by magnetic Wannier orbitals obtained by ab initio
calculations. It contrasts the ionic picture with a concrete example, and
presents a novel regime of the spin waves where the wavelength is comparable to
the spatial extent of the molecular orbitals. Our discovery brings important
insights into not only the magnetism of MnSi, but also a broad range of
magnetic quantum materials where structural symmetry, electron itinerancy and
correlations act in concert.

10 Aug 2021
The interaction between water and ions within droplets plays a key role in the chemical reactivity of atmospheric and man-made aerosols. Here we report direct computational evidence that in supercooled aqueous nanodroplets a lower density core of tetrahedrally coordinated water expels the cosmotropic ions to the denser and more disordered subsurface. In contrast, at room temperature, depending on the nature of the ion the radial distribution in the droplet core is nearly uniform or elevated towards the center. We analyze the spatial distribution of a single ion in terms of a reference electrostatic model. The energy of the system in the analytical model is expressed as the sum of the electrostatic and surface energy of a deformable droplet. The model predicts that the ion is subject to a harmonic potential centered at the droplet's center of mass. We name this effect "electrostatic confinement". The model's predictions are consistent with the simulation findings for a single ion at room temperature but not at supercooling. We anticipate this study to be the starting point for investigating the structure of supercooled (electro)sprayed droplets that are used to preserve the conformations of macromolecules originating from the bulk solution.

19 Apr 2023
University of Oslo Chinese Academy of Sciences
Chinese Academy of Sciences the University of Tokyo
the University of Tokyo Tsinghua University
Tsinghua University Westlake University
Westlake University Peking University
Peking University NVIDIA
NVIDIA Columbia UniversityXiamen UniversityNational University of Defense Technology
Columbia UniversityXiamen UniversityNational University of Defense Technology Rutgers University
Rutgers University Princeton UniversityEast China Normal University
Princeton UniversityEast China Normal University Flatiron InstituteHunan UniversityComenius UniversityBaidu IncQueen's University BelfastDP TechnologyAI for Science InstituteIndian Institute of Technology PalakkadSISSA - Scuola Internazionale Superiore di Studi Avanzati'
Ecole Polytechnique F
'ed
'erale de Lausanne
Flatiron InstituteHunan UniversityComenius UniversityBaidu IncQueen's University BelfastDP TechnologyAI for Science InstituteIndian Institute of Technology PalakkadSISSA - Scuola Internazionale Superiore di Studi Avanzati'
Ecole Polytechnique F
'ed
'erale de Lausanne
Chinese Academy of Sciencesthe University of TokyoTsinghua UniversityWestlake UniversityPeking UniversityNVIDIAColumbia UniversityXiamen UniversityNational University of Defense TechnologyRutgers UniversityPrinceton UniversityEast China Normal UniversityFlatiron InstituteHunan UniversityComenius UniversityBaidu IncQueen's University BelfastDP TechnologyAI for Science InstituteIndian Institute of Technology PalakkadSISSA - Scuola Internazionale Superiore di Studi Avanzati'
Ecole Polytechnique F
'ed
'erale de Lausanne
DeePMD-kit is a powerful open-source software package that facilitates molecular dynamics simulations using machine learning potentials (MLP) known as Deep Potential (DP) models. This package, which was released in 2017, has been widely used in the fields of physics, chemistry, biology, and material science for studying atomistic systems. The current version of DeePMD-kit offers numerous advanced features such as DeepPot-SE, attention-based and hybrid descriptors, the ability to fit tensile properties, type embedding, model deviation, Deep Potential - Range Correction (DPRc), Deep Potential Long Range (DPLR), GPU support for customized operators, model compression, non-von Neumann molecular dynamics (NVNMD), and improved usability, including documentation, compiled binary packages, graphical user interfaces (GUI), and application programming interfaces (API). This article presents an overview of the current major version of the DeePMD-kit package, highlighting its features and technical details. Additionally, the article benchmarks the accuracy and efficiency of different models and discusses ongoing developments.

18 Dec 2023
Researchers at Columbia University successfully created a Bose-Einstein Condensate of ground-state NaCs dipolar molecules, overcoming decades-long challenges in molecular cooling. They achieved this by implementing a novel dual-microwave shielding technique that engineers a purely repulsive intermolecular potential, suppressing both two- and three-body losses and enabling efficient evaporative cooling to temperatures as low as 6 nK with phase-space densities exceeding 1.
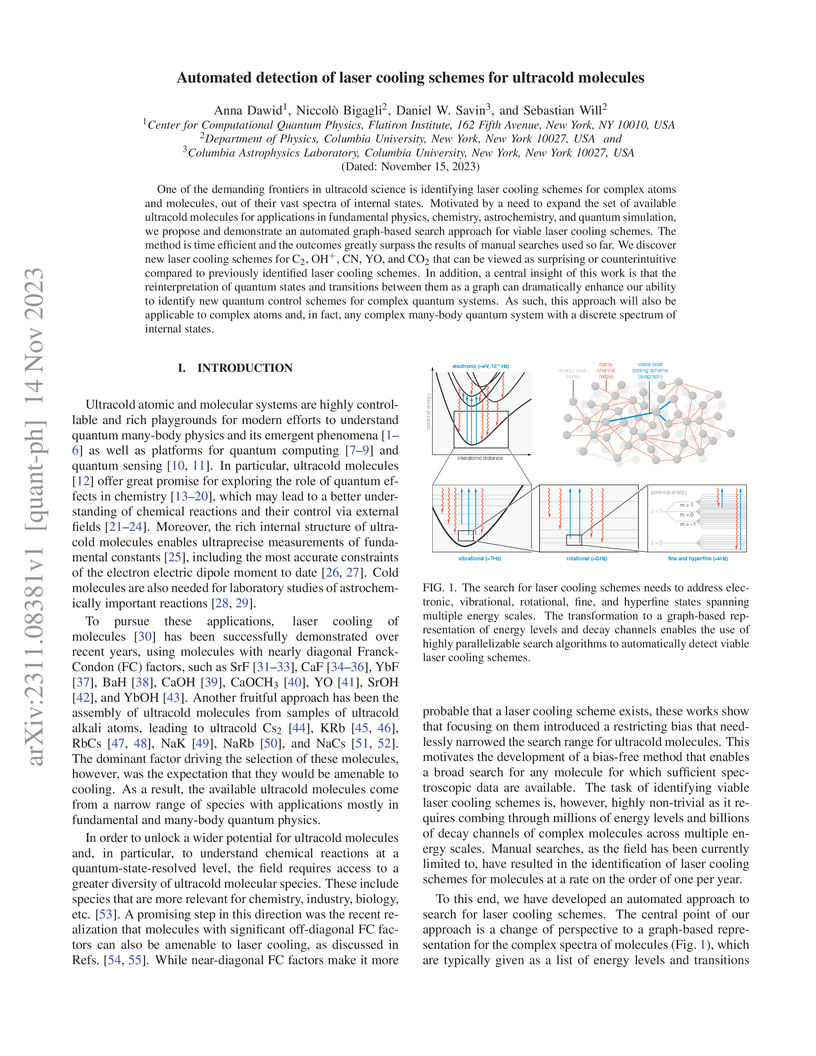
14 Nov 2023
One of the demanding frontiers in ultracold science is identifying laser cooling schemes for complex atoms and molecules, out of their vast spectra of internal states. Motivated by a need to expand the set of available ultracold molecules for applications in fundamental physics, chemistry, astrochemistry, and quantum simulation, we propose and demonstrate an automated graph-based search approach for viable laser cooling schemes. The method is time efficient and the outcomes greatly surpass the results of manual searches used so far. We discover new laser cooling schemes for C, OH, CN, YO, and CO that can be viewed as surprising or counterintuitive compared to previously identified laser cooling schemes. In addition, a central insight of this work is that the reinterpretation of quantum states and transitions between them as a graph can dramatically enhance our ability to identify new quantum control schemes for complex quantum systems. As such, this approach will also be applicable to complex atoms and, in fact, any complex many-body quantum system with a discrete spectrum of internal states.

20 May 2022
We investigate the depletion of single-electron states in small molecules under the influence of very short XUV pulses. In N, for a certain window of XUV energies around 50 eV, we observe a marked occupation inversion, i.e. a situation where depletion of the deepest bound valence electron state is much larger than for any other state. This represents a realistic mechanism which is able to cut, almost instantaneously, a hole into a deep lying state, a situation which is often assumed ad hoc in numerous theoretical studies of energetic ultrafast processes. This occupation inversion furthermore drives a dipole instability, i.e. a spontaneous reappearance of the dipole signal long after the laser pulse is over and the dipole signal has died out. The dipole signal that emerges from this instability can be identified as a particular low-energy structure in photo-electron spectra.

27 Sep 2024
The energy levels and transition rates of promising laser-cooling candidate thorium anion Th^- have been experimentally obtained in the current work. Three new excited states between the bound-bound electric dipole (E1) transition of Th^- are observed now, and their energy levels are determined to be 1,847(13), 3,166.8(59) and 3,666(12) cm^{-1}, respectively. Meanwhile, the lifetime of the upper state of that E1 transition is experimentally determined to be 30(2) microseconds, about 3 times shorter than the previous calculated result 86 microseconds, which makes Th^- the most promising candidate for laser cooling of negative ions. Furthermore, the lifetimes of two other short-lived odd-parity excited states of Th^- are also measured to be 59(4) and 53(3)microseconds, respectively.

09 Aug 2018
All possible non-isomorphic arrangements of 12 spheres kissing a central sphere (the Gregory-Newton problem) are obtained for the sticky-hard-sphere (SHS) model, and subsequently projected by geometry optimization onto a set of structures derived from an attractive Lennard-Jones (LJ) type of potential. It is shown that all 737 derived SHS contact graphs corresponding to the 12 outer spheres are (edge-induced) subgraphs of the icosahedral graph. The most widely used LJ(6,12) potential has only one minimum structure corresponding to the ideal icosahedron where the 12 outer spheres do not touch each other. The point of symmetry breaking away from the icosahedral symmetry towards the SHS limit is obtained for general LJ() potentials with exponents . Only if the potential becomes very repulsive in the short-range, determined by the LJ hard-sphere radius , symmetry broken solutions are observed.

17 Aug 2024
We introduce the elEmBERT model for chemical classification tasks. It is based on deep learning techniques, such as a multilayer encoder architecture. We demonstrate the opportunities offered by our approach on sets of organic, inorganic and crystalline compounds. In particular, we developed and tested the model using the Matbench and Moleculenet benchmarks, which include crystal properties and drug design-related benchmarks. We also conduct an analysis of vector representations of chemical compounds, shedding light on the underlying patterns in structural data. Our model exhibits exceptional predictive capabilities and proves universally applicable to molecular and material datasets. For instance, on the Tox21 dataset, we achieved an average precision of 96%, surpassing the previously best result by 10%.

07 May 2024
Hydrogen bonds are typically treated as sufficiently localized directional intermolecular bonds, in which dispersion and electrostatic contributions can be distinguished. However, being formed chiefly due to the overlapping of p orbitals of electronegative atoms, the corresponding electronic bonds are characterized by both {\sigma}- and {\pi}-kind binding, the former determining the directionality of bonds, while the latter, the coupling of molecules and the collective effects in H-bond networks. The latter contribution was never considered previously and is predetermined by overlapping pre-lone pair orbitals of oxygen atoms. This is manifested in the peculiarities of the electron density distribution, which are quantified based on the analysis of magnetic shielding tensors of oxygen and bridge hydrogen nuclei and illustrated by the shapes of cluster orbitals of water aggregates.

30 Jan 2020
Process of relaxation of Lennard-Jones cluster with an intrinsic pore was
investigated by molecular dynamics method for different phase states of an
initial cluster. Strong dependence of pore relaxation character on the initial
cluster phase state was demonstrated. It was shown that, for the initially
solid cluster, the system can reach metastable state where pore radius is fixed
at one of the discrete set of values. These values do not depend on initial
cluster temperature and size. Thus, it was demonstrated that, in a wide range
of cluster sizes, inside solid clusters "magic" pores can stabilize forming a
metastable state.

14 Sep 2018
Despite significant work on resource estimation for quantum simulation of
electronic systems, the challenge of preparing states with sufficient ground
state support has so far been largely neglected. In this work we investigate
this issue in several systems of interest, including organic molecules,
transition metal complexes, the uniform electron gas, Hubbard models, and
quantum impurity models arising from embedding formalisms such as dynamical
mean-field theory. Our approach uses a state-of-the-art classical technique for
high-fidelity ground state approximation. We find that easy-to-prepare single
Slater determinants such as the Hartree-Fock state often have surprisingly
robust support on the ground state for many applications of interest. For the
most difficult systems, single-determinant reference states may be
insufficient, but low-complexity reference states may suffice. For this we
introduce a method for preparation of multi-determinant states on quantum
computers.

21 Oct 2021
Electron removal in collisions of alpha particles with neon dimers is studied using an independent-atom-independent-electron model based on the semiclassical approximation of heavy-particle collision physics. The dimer is assumed to be frozen at its equilibrium bond length and collision events for the two ion-atom subsystems are combined in an impact parameter by impact parameter fashion for three mutually perpendicular orientations. Both frozen atomic target and dynamic response model calculations are carried out using the coupled-channel two-center basis generator method. We pay particular attention to inner-valence Ne() electron removal, which is associated with interatomic Coulombic decay (ICD), resulting in low-energy electron emission and dimer fragmentation. Our calculations confirm a previous experimental result at 150 keV/amu impact energy regarding the relative strength of ICD compared to direct electron emission. They further indicate that ICD is the dominant Ne + Ne fragmentation process below 10 keV/amu, suggesting that a strong low-energy electron yield will be observed in the ion-dimer system in a regime in which the creation of continuum electrons is a rare event in the ion-atom problem.

07 Jun 2024
We propose a method to trap polar molecules with the electrical force induced by the surface acoustic wave (SAW) on piezoelectric materials. In this approach, the electrical force is perpendicular to the moving direction of the polar molecules, and is used to control the positions of trapped polar molecules in the direction orthogonal to the acoustic transmission. By virtue of an external electrical force, the SAW-induced electrical field can trap the polar molecules into single-layer or multi-layer lattices. The arrangement of molecules can affect the binding energy and localization of the molecule array. Then the one- or two-dimensional trapped polar molecule arrays can be used to construct the Bose-Hubbard (BH) model, whose energy and dynamics are affected by the localizations of the trapped molecules. We find that the phase transitions between the superfluid and Mott insulator based on trapped polar molecule BH model can be modulated by the SAW induced electrical potential.

09 Mar 2024
The advancement of liquid phase electron beam induced deposition has enabled an effective direct-write approach for functional nanostructure synthesis with the possibility of three-dimensional control of morphology. For formation of a metallic solid phase, the process employs ambient temperature, beam-guided, electrochemical reduction of precursor cations resulting in rapid formation of structures, but with challenges for retention of resolution achievable via slower electron beam approaches. The possibility of spatial control of redox pathways via the use of water-ammonia solvents has opened new avenues for improved nanostructure resolution without sacrificing the growth rate. We find that ammonia concentration locally modulates reaction kinetics, altering the balance between reducing and oxidizing species, leading to distinct deposition outcomes. The key effect is an 'electrochemical lensing', achieved at an optimum ammonia concentration, in which a tightly confined and highly reducing environment is created locally to enable high resolution, rapid beam-directed nanostructure growth. We demonstrate this unique approach to high resolution synthesis through a combination of analysis and experiment.

01 Feb 2017
A novel system containing nanoporous materials and carbonate ions is proposed, which is capable to capture CO2 from ambient air simply by controlling the amount of water (humidity) in the system. The system absorbs CO2 from the air when the surrounding is dry, whereas desorbs CO2 when wet. A design of such a CO2 absorption/desorption system is investigated in this paper using molecular dynamics and quantum mechanics simulations, and also verified by experiments. Its working mechanism is revealed as the reduction of free energy of the carbonate ion hydrolysis with the decrease of the number of water molecules in confined nano-pores. The influences of pore size, spacing of cations, surface hydrophobicity and temperature on CO2 capture efficiency are elucidated. The study may help to design an efficient direct air capture system and contribute to the negative carbon emission.

01 Apr 2022
We have designed a Euler approximation Monte-Carlo code for the calculation of the ionization of atomic hydrogen by protons with energies between 0.05 and 1.0 MeV. This code is used to calculate the total ionization cross sections s(E), the angular distribution double differential cross sections d2s/ (dW - dE) for electrons with energies of 150 eV ionized by 300 keV protons and the energy distribution d2s/ (dW - dE) for the ionized electrons emitted at 10o by protons of energy 300 keV. Our calculations are compared to experimental values of these quantities as well as independent calculations. We find excellent agreement of our calculated cross section and reasonable agreement of our calculated double differential cross section with independent calculations and measurements.

22 May 2024
 ETH ZurichKansas State University
ETH ZurichKansas State University Argonne National LaboratoryDeutsches Elektronen-Synchrotron DESYPaul Scherrer InstitutLa Trobe UniversityUniversit\"at HamburgAtomic Energy and Alternative Energies CommissionUniversity of Nebraska-LincolnEuropean XFELNara Women’s UniversityoptiX fab GmbHTechnische Universitat¨ Berlin["École Polytechnique Fédérale de Lausanne"]
Argonne National LaboratoryDeutsches Elektronen-Synchrotron DESYPaul Scherrer InstitutLa Trobe UniversityUniversit\"at HamburgAtomic Energy and Alternative Energies CommissionUniversity of Nebraska-LincolnEuropean XFELNara Women’s UniversityoptiX fab GmbHTechnische Universitat¨ Berlin["École Polytechnique Fédérale de Lausanne"]Because of their high photon flux, X-ray free-electron lasers (FEL) allow to resolve the structure of individual nanoparticles via coherent diffractive imaging (CDI) within a single X-ray pulse. Since the inevitable rapid destruction of the sample limits the achievable resolution, a thorough understanding of the spatiotemporal evolution of matter on the nanoscale following the irradiation is crucial. We present a technique to track X-ray induced structural changes in time and space by recording two consecutive diffraction patterns of the same single, free-flying nanoparticle, acquired separately on two large-area detectors opposite to each other, thus examining both the initial and evolved particle structure. We demonstrate the method at the extreme ultraviolet (XUV) and soft X-ray Free-electron LASer in Hamburg (FLASH), investigating xenon clusters as model systems. By splitting a single XUV pulse, two diffraction patterns from the same particle can be obtained. For focus intensities of about we observe still largely intact clusters even at the longest delays of up to 650 picoseconds of the second pulse, indicating that in the highly absorbing systems the damage remains confined to one side of the cluster. Instead, in case of five times higher flux, the diffraction patterns show clear signatures of disintegration, namely increased diameters and density fluctuations in the fragmenting clusters. Future improvements to the accessible range of dynamics and time resolution of the approach are discussed.

06 Mar 2024
Correlated rotational alignment spectroscopy correlates observables of ultrafast gas-phase spectroscopy with high-resolution, broad-band rotational Raman spectra. This article reviews the measurement principle of CRASY, existing implementations for mass-correlated measurements, and the potential for future developments. New spectroscopic capabilities are discussed in detail: Signals for individual sample components can be separated even in highly heterogeneous samples. Isotopologue rotational spectra can be observed at natural isotope abundance. Fragmentation channels are readily assigned in molecular and cluster mass spectra. And finally, rotational Raman spectra can be measured with sub-MHz resolution, an improvement of several orders-of-magnitude as compared to preceding experiments.
There are no more papers matching your filters at the moment.
