
19 Sep 2025

Fluorite ferroelectrics are exciting candidates for next-generation non-volatile memory devices because their unique ferroelectric mechanism, which arises from unconventional oxygen displacements, permits ferroelectricity with minimal thickness constraints. However, the polarisation switching mechanism remains the subject of intense debate due to a limited understanding of the atomic-scale dynamics which are extremely challenging to detect and measure. Here, we observe directly the polarisation switching pathways by visualising oxygen site dynamics in ZrO2 and Hf0.5Zr0.5O2 freestanding membranes using an advanced atomic-column imaging technique-optimum bright-field scanning transmission electron microscopy. We observe that the 180- and 90-degree polarisation pathways involve different nonpolar intermediate states with distinct spatial scales. Coupled with density functional theory, we also reveal how different cation species in fluorite oxides impact the accessible polarisation switching pathways. Our atomic-level insights into the polarisation switching dynamics open new avenues for the advanced engineering of fluorite ferroelectric materials and resulting memory devices.

02 Feb 2022

Max-value entropy search (MES) is one of the state-of-the-art approaches in
Bayesian optimization (BO). In this paper, we propose a novel variant of MES
for constrained problems, called Constrained MES via Information lower BOund
(CMES-IBO), that is based on a Monte Carlo (MC) estimator of a lower bound of a
mutual information (MI). Unlike existing studies, our MI is defined so that
uncertainty with respect to feasibility can be incorporated. We derive a lower
bound of the MI that guarantees non-negativity, while a constrained counterpart
of conventional MES can be negative. We further provide theoretical analysis
that assures the low-variability of our estimator which has never been
investigated for any existing information-theoretic BO. Moreover, using the
conditional MI, we extend CMES-IBO to the parallel setting while maintaining
the desirable properties. We demonstrate the effectiveness of CMES-IBO by
several benchmark functions and real-world problems.

13 Mar 2024
A computer algorithm to search symmetries of crystal structures as implemented in the \texttt{spglib} code is described. An iterative algorithm is employed to robustly identify space group types tolerating a certain amount of distortion in the crystal structures. The source code is distributed under the 3-Clause BSD License, a permissive open-source software license. This paper focuses on the algorithm for identifying the space group symmetry of the crystal structures.

08 Jan 2019
Many rotational invariants for crystal structure representations have been
used to describe the structure-property relationship by machine learning. The
machine learning interatomic potential (MLIP) is one of the applications of
rotational invariants, which provides the relationship between the energy and
the crystal structure. Since the MLIP requires the highest accuracy among
machine learning estimations of the structure-property relationship, the
enumeration of rotational invariants is useful for constructing MLIPs with the
desired accuracy. In this study, we introduce high-order linearly independent
rotational invariants up to the sixth order based on spherical harmonics and
apply them to linearized MLIPs for elemental aluminum. A set of rotational
invariants is derived by the general process of reducing the Kronecker products
of irreducible representations (Irreps) for the SO(3) group using a
group-theoretical projector method. A high predictive power for a wide range of
structures is accomplished by using high-order invariants with low-order
invariants equivalent to pair and angular structural features.

27 Dec 2016

Lattice thermal conductivities (LTCs) of \alpha-, \beta-, and
\gamma-SiN single crystals are investigated from ab initio anharmonic
lattice dynamics, within the single-mode relaxation-time approximation of the
linearized phonon Boltzmann transport equation. At a temperature of 300 K, a
\kappa of 70 and a \kappa of 98 (in units of WmK)
are obtained for \alpha-SiN. For \beta-SiN, \kappa and
\kappa are found to be 71 and 194, respectively, which are consistent
with the reported experimental values of 69 and 180 for individual
\beta-SiN grains in a ceramic. The theoretical \kappa values of
\alpha- and \beta-SiN are comparable, while the \kappa value of
\beta-SiN is almost twice that of \alpha-SiN, which
demonstrates the very large anisotropy in the LTC of the \beta phase. It is
found that the large anisotropy in the LTC of \beta-SiN was caused by
the elongated Brillouin zone along the c* axis, where the acoustic phonons
mostly contribute to LTC and have large group velocities even near the
Brillouin zone boundary. The LTC of \gamma-SiN is 81, which is as small
as that of \alpha-SiN, although \gamma-SiN has much larger
elastic constants. This means that elastic constants are not always a good
indicator of LTC. We show that knowing the detailed distributions of both the
group velocities and phonon lifetimes in the Brillouin zones is important for
characterizing the LTC of the three phases.

18 Jan 2018
 the University of Tokyo
the University of Tokyo Kyoto UniversityNagoya Institute of TechnologyJSTNational Institute for Materials ScienceRIKEN Center for Advanced Intelligence ProjectGifu UniversityJapan Fine Ceramics CenterCenter for Materials Research by Information IntegrationCenter for Elements Strategy Initiative for Structure Materials
Kyoto UniversityNagoya Institute of TechnologyJSTNational Institute for Materials ScienceRIKEN Center for Advanced Intelligence ProjectGifu UniversityJapan Fine Ceramics CenterCenter for Materials Research by Information IntegrationCenter for Elements Strategy Initiative for Structure MaterialsWe propose a machine-learning method for evaluating the potential barrier
governing atomic transport based on the preferential selection of dominant
points for the atomic transport. The proposed method generates numerous random
samples of the entire potential energy surface (PES) from a probabilistic
Gaussian process model of the PES, which enables defining the likelihood of the
dominant points. The robustness and efficiency of the method are demonstrated
on a dozen model cases for proton diffusion in oxides, in comparison with a
conventional nudge elastic band method.

19 Mar 2020
Ion-conducting solid electrolytes are widely used for a variety of purposes.
Therefore, designing highly ion-conductive materials is in strongly demand.
Because of advancement in computers and enhancement of computational codes,
theoretical simulations have become effective tools for investigating the
performance of ion-conductive materials. However, an exhaustive search
conducted by theoretical computations can be prohibitively expensive. Further,
for practical applications, both dynamic conductivity as well as static
stability must be satisfied at the same time. Therefore, we propose a
computational framework that simultaneously optimizes dynamic conductivity and
static stability; this is achieved by combining theoretical calculations and
the Bayesian multi-objective optimization that is based on the Pareto
hyper-volume criterion. Our framework iteratively selects the candidate
material, which maximizes the expected increase in the Pareto hyper-volume
criterion; this is a standard optimality criterion of multi-objective
optimization. Through two case studies on oxygen and lithium diffusions, we
show that ion-conductive materials with high dynamic conductivity and static
stability can be efficiently identified by our framework.

06 Feb 2014
A combination of systematic density functional theory (DFT) calculations and
machine learning techniques has a wide range of potential applications. This
study presents an application of the combination of systematic DFT calculations
and regression techniques to the prediction of the melting temperature for
single and binary compounds. Here we adopt the ordinary least-squares
regression (OLSR), partial least-squares regression (PLSR), support vector
regression (SVR) and Gaussian process regression (GPR). Among the four kinds of
regression techniques, the SVR provides the best prediction. In addition, the
inclusion of physical properties computed by the DFT calculation to a set of
predictor variables makes the prediction better. Finally, a simulation to find
the highest melting temperature toward the efficient materials design using
kriging is demonstrated. The kriging design finds the compound with the highest
melting temperature much faster than random designs. This result may stimulate
the application of kriging to efficient materials design for a broad range of
applications.
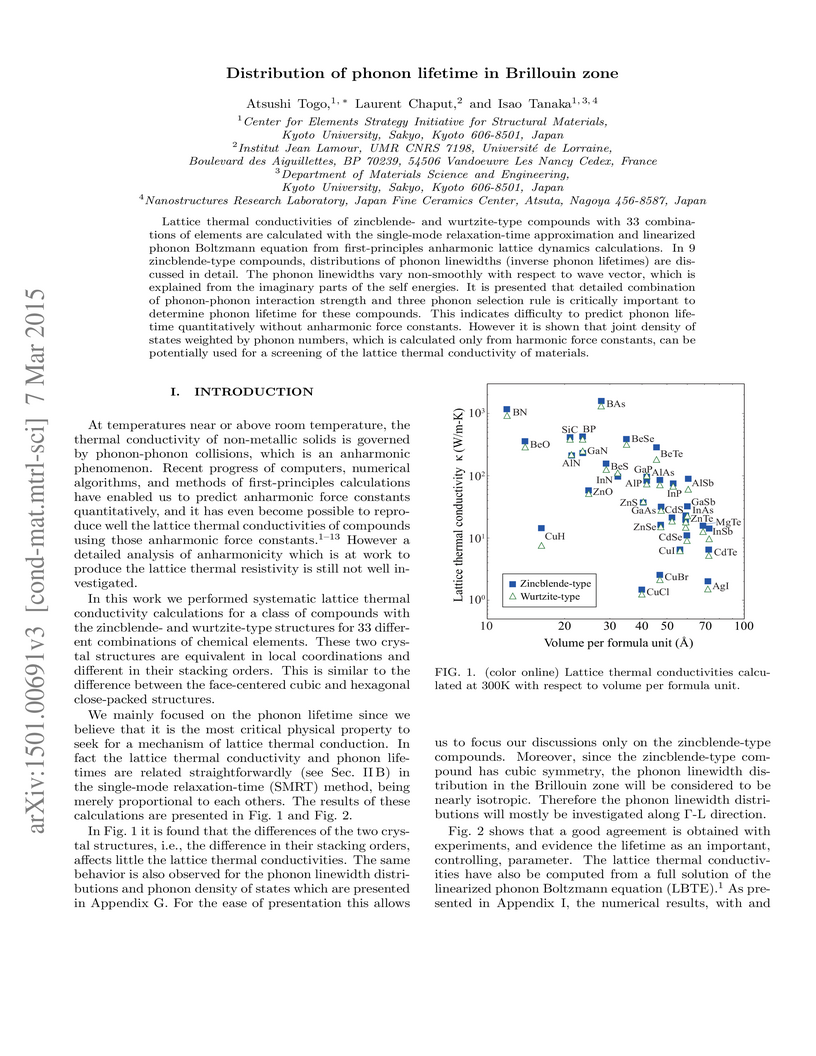
07 Mar 2015
Lattice thermal conductivities of zincblende- and wurtzite-type compounds
with 33 combinations of elements are calculated with the single-mode
relaxation-time approximation and linearized phonon Boltzmann equation from
first-principles anharmonic lattice dynamics calculations. In 9 zincblende-type
compounds, distributions of phonon linewidths (inverse phonon lifetimes) are
discussed in detail. The phonon linewidths vary non-smoothly with respect to
wave vector, which is explained from the imaginary parts of the self energies.
It is presented that detailed combination of phonon-phonon interaction strength
and three phonon selection rule is critically important to determine phonon
lifetime for these compounds. This indicates difficulty to predict phonon
lifetime quantitatively without anharmonic force constants. However it is shown
that joint density of states weighted by phonon numbers, which is calculated
only from harmonic force constants, can be potentially used for a screening of
the lattice thermal conductivity of materials.

22 Jun 2015
Compounds of low lattice thermal conductivity (LTC) are essential for seeking
thermoelectric materials with high conversion efficiency. Some strategies have
been used to decrease LTC. However, such trials have yielded successes only
within a limited exploration space. Here we report the virtual screening of a
library containing 54,779 compounds. Our strategy is to search the library
through Bayesian optimization using for the initial data the LTC obtained from
first-principles anharmonic lattice dynamics calculations for a set of 101
compounds. We discovered 221 materials with very low LTC. Two of them have even
an electronic band gap < 1 eV, what makes them exceptional candidates for
thermoelectric applications. In addition to those newly discovered
thermoelectric materials, the present strategy is believed to be powerful for
many other applications in which chemistry of materials are required to be
optimized.

21 Dec 2016
The representations of a compound, called "descriptors" or "features", play an essential role in constructing a machine-learning model of its physical properties. In this study, we adopt a procedure for generating a systematic set of descriptors from simple elemental and structural representations. First it is applied to a large dataset composed of the cohesive energy for about 18000 compounds computed by density functional theory (DFT) calculation. As a result, we obtain a kernel ridge prediction model with a prediction error of 0.041 eV/atom, which is close to the "chemical accuracy" of 1 kcal/mol (0.043 eV/atom). The procedure is also applied to two smaller datasets, i.e., a dataset of the lattice thermal conductivity (LTC) for 110 compounds computed by DFT calculation and a dataset of the experimental melting temperature for 248 compounds. We examine the performance of the descriptor sets on the efficiency of Bayesian optimization in addition to the accuracy of the kernel ridge regression models. They exhibit good predictive performances.

30 Nov 2021
The advanced data structure of the zero-suppressed binary decision diagram (ZDD) enables us to efficiently enumerate nonequivalent substitutional structures. Not only can the ZDD store a vast number of structures in a compressed manner, but also can a set of structures satisfying given constraints be extracted from the ZDD efficiently. Here, we present a ZDD-based efficient algorithm for exhaustively searching for special quasirandom structures (SQSs) that mimic the perfectly random substitutional structure. We demonstrate that the current approach can extract only a tiny number of SQSs from a ZDD composed of many substitutional structures (>). As a result, we find SQSs that are optimized better than those proposed in the literature. A series of SQSs should be helpful for estimating the properties of substitutional solid solutions. Furthermore, the present ZDD-based algorithm should be useful for applying the ZDD to the other structure enumeration problems.

29 Dec 2021
Zirconia ceramics have been known as a structural material with high fracture
toughness and strength since Garvie et al. discovered phase transformation
toughening in 1975 [1]. Although these mechanical properties are the most
excellent among the advanced ceramics, the toughness has not yet reached the
level of metallic materials. Here, we demonstrate for the first time that 2.9
mass% Y2O3-stabilized ZrO2s doped with Al2O3 greatly exceed the toughness of
conventional zirconia ceramics which is comparable to those of metallic
materials, even with slightly higher strength. The excellent mechanical
properties in the proposed ceramic materials will be useful for further
expanding the application of advanced ceramics to many engineering fields.

28 Apr 2023
Using first-principles calculations we examine the crystal structures and phase transitions of nitride perovskite LaWN. Lattice dynamics calculations indicate that the ground-state structure belongs to space group . Two competitive phase transition pathways are identified which are characterized by symmetry-adapted distortion modes. The results suggest that LaWN should be an excellent ferroelectric semiconductor: its large spontaneous polarization of around 61 C/cm is comparable to that of PbTiO, and its band gap is about 1.72 eV. Ferroelectricity is found to result from the \emph{B}-site instability driven by hybridization between W-5 and N-2 orbitals. These properties make LaWN an attractive candidate material for use in ferroelectric memory devices and photovoltaic cells.

21 Oct 2022

The addition of artificial pinning centers has led to an impressive increase
in critical current density () in a superconductor, enabling
record-breaking all-superconducting magnets and other applications.
has reached - , where is the depairing
current density, and the numerical factor depends on the pinning optimization.
By modifying and/or , the penetration depth and coherence
length, respectively, we can increase . For
(YGd)BaCuO ((Y,Gd)123) we achieve this by
controlling the carrier density, which is related to and . We
also tune and by controlling the chemical pressure in the
Fe-based superconductors, BaFe(AsP) films. The variation of
and leads to an intrinsic improvement of , via
, obtaining extremely high values of of MA/cm
and MA/cm at K, consistent with an enhancement of
of a factor of for both incoherent nanoparticle-doped (Y,Gd)123 coated
conductors (CCs) and BaFe(AsP) films, showing that this new
material design is useful to achieving high critical current densities for a
wide array of superconductors. The remarkably high vortex-pinning force in
combination with this thermodynamic and pinning optimization route for the
(Y,Gd)123 CCs reached TN/m at K and 18 T (${\bf
H}\parallel c$), the highest values ever reported in any superconductor.

13 Jan 2023
Scientific simulation codes are public property sustained by the community.
Modern technology allows anyone to join scientific software projects, from
anywhere, remotely via the internet.
The phonopy and phono3py codes are widely used open source phonon calculation
codes. This review describes a collection of computational methods and
techniques as implemented in these codes and shows their implementation
strategies as a whole, aiming to be useful for the community. Some of the
techniques presented here are not limited to phonon calculations and may
therefore be useful in other area of condensed matter physics.

28 Apr 2023
Using first-principles calculations we examine the crystal structures and
phase transitions of nitride perovskite LaWN. Lattice dynamics calculations
indicate that the ground-state structure belongs to space group . Two
competitive phase transition pathways are identified which are characterized by
symmetry-adapted distortion modes. The results suggest that LaWN
should be an excellent ferroelectric semiconductor: its large spontaneous
polarization of around 61 C/cm is comparable to that of PbTiO, and
its band gap is about 1.72 eV. Ferroelectricity is found to result from the
\emph{B}-site instability driven by hybridization between W-5 and N-2
orbitals. These properties make LaWN an attractive candidate material for
use in ferroelectric memory devices and photovoltaic cells.

28 Aug 2014
The use of a special quasirandom structure (SQS) is a rational and efficient way to approximate random alloys. A wide variety of physical properties of metallic and semiconductor random alloys have been successfully estimated by a combination of an SQS and density functional theory (DFT) calculation. Here, we investigate the application of an SQS to the ionic multicomponent systems with configurations of heterovalent ions, including point-charge lattices, MgAlO and ZnSnP. It is found that the physical properties do not converge with the supercell size of the SQS. This is ascribed to the fact that the correlation functions of long-range clusters larger than the period of the supercell are not optimized in the SQS. However, we demonstrate that the physical properties of the perfectly disordered structure can be estimated by linear extrapolation using the inverse of the supercell size.

31 Jan 2025

Functionalities in crystalline materials are determined by 3-dimensional collective interactions of atoms. The confinement of dimensionality in condensed matter provides an exotic research direction to understand the interaction of atoms, thus can be used to tailor or create new functionalities in material systems. In this study, a 2-dimensional transition metal oxide monolayer is constructed inside complex oxide heterostructures based on the theoretical predictions. The electrostatic boundary conditions of oxide monolayer in the heterostructure is carefully designed to tune the chemical, electronic, and magnetic states of oxide monolayer. The challenge of characterizing such an oxide monolayer is overcome by a combination of transmission electron microscopy, x-ray absorption spectroscopy, cross-sectional scanning tunneling microscopy, and electrical transport measurements. An intriguing metal-insulator transition associated with a magnetic transition is discovered in the MnO2 monolayer. This study paves a new route to understand the confinement of dimensionality and explore new intriguing phenomena in condensed matters.

01 Jul 2025

The electromagnetic properties of type-II superconductors depend on vortices-magnetic flux lines whose motion introduces dissipation that can be mitigated by pinning from material defects. The material disorder landscape is tuned by the choice of materials growth technique and incorporation of impurities that serve as vortex pinning centers. For example, metal organic deposition (MOD) and pulsed laser deposition (PLD) produce high-quality superconducting films with uncorrelated versus correlated disorder, respectively. Here, we study vortex dynamics in PLD-grown EuBa2Cu3Oy films containing varying concentrations of BaHfO3 inclusions and compare our results with those of MOD-grown (Y,Gd)Ba2Cu3Oy films. Despite both systems exhibiting behavior consistent with strong pinning theory, which predicts the critical current density J_c based on vortex trapping by randomly distributed spherical inclusions, we find striking differences in the vortex dynamics owing to the correlated versus uncorrelated disorder. Specifically, we find that the EuBa2Cu3Oy films grown without inclusions exhibit surprisingly slow vortex creep, comparable to the slowest creep rates achieved in (Y,Gd)Ba2Cu3Oy films containing high concentrations of BaHfO3. Whereas adding inclusions to (Y,Gd)Ba2Cu3Oy is effective in slowing creep, BaHfO3 increases creep in EuBa2Cu3Oy even while concomitantly improving J_c. Lastly, we find evidence of variable range hopping and that J_c is maximized at the BaHfO3 concentration that hosts a vortex or Bose glass state.
There are no more papers matching your filters at the moment.
