
09 Dec 2025
We present a fully analytical integration of the Maxwell stress tensor and derive exact relations for interparticle forces in systems of multiple dielectric spheres immersed in a polarizable ionic solvent, within the framework of the linearized Poisson--Boltzmann theory. Building upon the screening-ranged (in ascending orders of Debye screening) expansions of the potentials developed and rigorously analyzed in the accompanying works \cite{supplem_pre,supplem_pre_math,supplem_prl}, we construct exact screening-ranged many-body expansions for electrostatic forces in explicit analytical form. These results establish a rigorous foundation for evaluating screened electrostatic interactions in complex particle systems and provide direct analytical connections to, and systematic improvements upon, various earlier approximate or limited-case formulations available in the literature, both at zero and finite ionic strength.

09 Dec 2025
We present an analytical many-body formalism for systems of spherical particles carrying arbitrary free charge distributions and interacting in a polarizable electrolyte solution, that we model within the linearized Poisson--Boltzmann framework. Building on the detailed spectral analysis of the associated nonstandard Neumann--Poincaré-type operators developed in our companion study~\cite{supplem_pre_math}, we construct exact explicit expansions of the electrostatic potential and energy in ascending orders of Debye screening thereby obtaining systematic "screening-ranged" series for potentials and energies. These screening-ranged expansions provide a unified and tractable description of many-body electrostatics. We demonstrate the versatility of the approach by showing how it generalizes and improves upon both classical and modern methods, enabling rigorous treatment of heterogeneously charged systems (such as Janus particles) and accurate modeling of higher-order phenomena (such as asymmetric dielectric screening, opposite-charge repulsion, like-charge attraction) as well as yielding many-body generalizations to analytical explicit results previously known only in the two-body setting.
09 Dec 2025
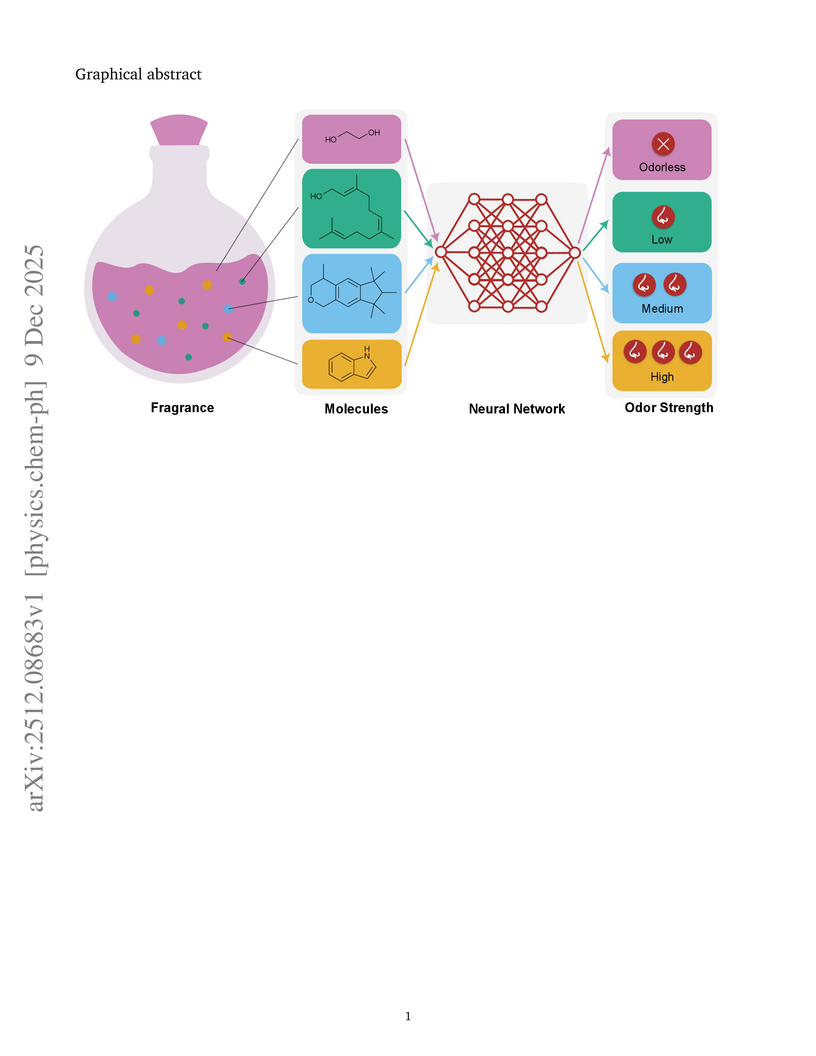
Predicting olfactory perception directly from molecular structure is central to fragrance design that plays a role in a wide range of industries, such as perfumery, food and beverage, and health care. Among olfactory attributes, odor strength is a key factor in shaping odor perception, but its modeling has been impeded by scarce and fragmented intensity data. In this work, we introduce an ordinal odor strength data set of over 2,000 molecules by integrating two different public sources, mapping structures to odorless, low, medium, and high categories. Across several molecular encodings and supervised learning algorithms we compared different prediction strategies. Dimensionality reduction and SHAP analysis identifies molecular size, polarity, ring features, and branching as primary drivers, consistent with mass-transport constraints on volatility, sorption, and receptor access. This scalable ordinal framework enables reliable odor-strength estimation for novel molecules and provides a foundation for in silico fragrance design.

09 Dec 2025
The interaction of particles in an electrolytic medium can be calculated by solving the Poisson equation inside the solutes and the linearized Poisson--Boltzmann equation in the solvent, with suitable boundary conditions at the interfaces. Analytical approaches often expand the potentials in spherical harmonics, relating interior and exterior coefficients and eliminating some coefficients in favor of others, but a rigorous spectral analysis of the corresponding formulations is still lacking. Here, we introduce composite many-body Neumann--Poincaré-type operators and prove that they are compact with spectral radii strictly less than one. These results provide the foundation for systematic screening-ranged expansions, in powers of the Debye screening parameters, of electrostatic potentials, interaction energies, and forces, and establish the analytical framework for the accompanying works~\cite{supplem_prl,supplem_pre,supplem_pre_force}.

08 Dec 2025

Coarse-grained (CG) modeling enables molecular simulations to reach time and length scales inaccessible to fully atomistic methods. For classical CG models, the choice of mapping, that is, how atoms are grouped into CG sites, is a major determinant of accuracy and transferability. At the same time, the emergence of machine learning potentials (MLPs) offers new opportunities to build CG models that can in principle learn the true potential of the mean force for any mapping. In this work, we systematically investigate how the choice of mapping influences the representations learned by equivariant MLPs by studying liquid hexane, amino acids, and polyalanine. We find that when the length scales of bonded and nonbonded interactions overlap, unphysical bond permutations can occur. We also demonstrate that correctly encoding species and maintaining stereochemistry are crucial, as neglecting either introduces unphysical symmetries. Our findings provide practical guidance for selecting CG mappings compatible with modern architectures and guide the development of transferable CG models.

07 Dec 2025
The launch of the James Webb Space Telescope (JWST) has delivered high-quality atmospheric observations and expanded the known chemical inventory of exoplanetary atmospheres, opening new avenues for atmospheric chemistry modeling to interpret these data. Here, we present XODIAC, a fast, GPU-accelerated, one-dimensional photochemical model with a built-in equilibrium chemistry solver, an updated thermochemical database, and three chemical reaction networks. This framework enables comparative atmospheric chemistry studies, including the newly developed XODIAC-2025 network, a state-of-the-art C-H-O-N-P-S-Metals network, linking 594 species through 7,720 reactions. The other two are existing, publicly available C-H-O-N-S and C-H-O-N-S-Metals networks, from the established photochemical models VULCAN and ARGO, respectively, which are commonly used in the community. The XODIAC model has been rigorously benchmarked on the well-studied hot Jupiter HD 189733 b, with results compared against these two models. Benchmarking shows excellent agreement and demonstrates that, when the same chemical network and initial conditions are used, the numerical scheme for solving atmospheric chemistry does not significantly affect the results. We also revisited the atmospheric chemistry of HD 189733 b and performed a comparative analysis across the three networks. Sulfur chemistry shows the least variation across networks, carbon chemistry shows slightly more, and phosphorus chemistry varies the most, primarily due to the introduction of unique PHO and PN pathways comprising 390 reactions in the XODIAC-2025 network. These findings highlight XODIAC's capability to advance exoplanetary atmospheric chemistry and provide a robust framework for comparative exoplanetology.

08 Dec 2025
Rohit Goswami, affiliated with EPFL and TurtleTech ehf., introduced a two-dimensional RMSD projection method for visualizing and validating reaction pathways in computational chemistry. This method maps high-dimensional reaction trajectories onto a 2D surface based on intrinsic geometric distances, allowing for robust convergence diagnosis, clear landscape topology visualization, and nuanced comparison of different potential energy surfaces.

10 Dec 2025
We present an exact many-body framework for electrostatic interactions among arbitrarily charged spheres in an electrolyte, modeled by the linearized Poisson--Boltzmann equation. Building on a spectral analysis of nonstandard Neumann--Poincaré-type operators introduced in a companion mathematical work~\cite{supplem_pre_math}, we construct convergent screening-ranged series for the potential, interaction energy, and forces, where each term is associated with a well-defined Debye--Hückel screening order and can be obtained evaluating an analytical expression rather than numerically solving an infinitely dimensional linear system. This formulation unifies and extends classical and recent approaches, providing a rigorous basis for electrostatic interactions among heterogeneously charged particles (including Janus colloids) and yielding many-body generalizations of analytical closed-form results previously available only for two-body systems. The framework captures and clarifies complex effects such as asymmetric dielectric screening, opposite-charge repulsion, and like-charge attraction, which remain largely analytically elusive in existing treatments. Beyond its fundamental significance, the method leads to numerically efficient schemes, offering a versatile tool for modeling colloids and soft/biological matter in electrolytic solution.

10 Dec 2025
Accurate prediction of the frequency response of quantum dots under electromagnetic radiation is essential for investigating absorption spectra, excitonic effects, and nonlinear optical behavior in quantum dots and semiconductor nanoparticles. The polarization propagator provides a rigorous framework for evaluating these properties, but its construction is computationally demanding. Challenges arise from the level of electron correlation, the size of the excitonic basis, and the cost of evaluating two-electron integrals. This work addresses these difficulties by developing first- and second-order frequency-dependent polarization propagator calculations for PbS and CdS quantum dots. The propagator is formulated using the electron propagator approach and expressed as the resolvent of the Hamiltonian superoperator. Light-matter interaction is treated using the dipole approximation and represented in a particle-hole excitation operator basis. The correlated ground state is treated at the MP2 level, and all response-matrix terms up to second order in the fluctuating potential are included. A frequency-dependent inverse Krylov subspace method is derived and combined with the folded-spectrum technique to isolate excitation energies within a chosen frequency window. This strategy avoids full diagonalization of the response matrix and significantly reduces computational cost for large systems. The method is implemented in a matrix-free manner in which no explicit response matrix is assembled, and all operations rely on matrix-vector products. UV-VIS excitation spectra of PbS and CdS quantum dots were computed, demonstrating that the inverse Krylov subspace projection approach provides an efficient and accurate approximation for excitation spectra when full diagonalization is computationally prohibitive.

10 Dec 2025
The association and dissociation of ion pairs in water are fundamental to physical chemistry, yet their reaction coordinates are complex, involving not only interionic distance but also solvent-mediated hydration structures. These processes are often represented by free-energy landscapes constructed from collective variables (CVs), such as interionic distance and water bridging structures; however, it remains uncertain whether such representations reliably capture the transition pathways between the two associated and dissociated states. In this study, we employ deep learning to identify reaction coordinates for NaCl ion pair association and dissociation in water, using the committor as a quantitative measure of progress along the transition pathway through the transition state. The solvent environment surrounding the ions is encoded through descriptors based on atom-centered symmetry functions (ACSFs), which serve as input variables for the neural network. In addition, an explainable artificial intelligence technique is applied to identify ACSFs that contribute to the reaction coordinate. A comparative analysis of their correlation with CVs representing water bridging structures, such as interionic water density and the number of water molecules coordinating both ions, further provides a molecular-level interpretation of the ion association-dissociation mechanism in water.

10 Dec 2025
Belite -- dicalcium silicate CaSiO -- is a main constituent of low-carbon cement. In this work, we study several terminations of the (010) surface of -belite, its most stable polymorph, by molecular dynamics simulations. The energies and forces are provided by a high-dimensional neural network potential trained to density functional theory data. Water can interact in molecular form as well as dissociatively with the investigated interfaces, and the degree of dissociation is determined primarily by the protonation of SiO groups accessible at the surface. A major part of the simultaneously formed hydroxide ions is adsorbed at surface calcium atoms, whose octahedral coordination spheres are completed by additional water molecules. The T3 termination, which is most stable in vacuum, shows only little reactivity in water. For the only slightly less stable T2 termination, however, two distinct types of surface defects are observed. The type I defect is even stable in vacuum and leads to a reconstruction of the entire surface, while the type II defect is only found in the presence of water. Overall, our results suggest that a variety of structures may be formed at the CaSiO(010) surface, which are stabilized in the presence of water.

05 Dec 2025
A major challenge in light-matter simulations is bridging the disparate time and length scales of electrodynamics and molecular dynamics. Current computational approaches often rely on heuristic approximations of either the electromagnetic (EM) or material component, hindering the exploration of complex light-matter systems. Herein, MaxwellLink -- a modular, open-source Python framework -- is developed for the massively parallel, self-consistent propagation of classical EM fields interacting with a large heterogeneous molecular ensemble. The package utilizes a robust TCP/UNIX socket interface to couple EM solvers with a wide range of external molecular drivers. This decoupled architecture allows users to seamlessly switch between levels of theory of either the EM solver or molecules without modifying the counterpart. Crucially, MaxwellLink supports EM solvers spanning from single-mode cavities to full-feature three-dimensional finite-difference time-domain (FDTD) engines, and molecules described by multilevel open quantum systems, force-field and first-principles molecular dynamics, and nonadiabatic real-time Ehrenfest dynamics. Benefiting from the socket-based design, the EM engine and molecular drivers scale independently across multiple high-performance computing (HPC) nodes, facilitating large-scale simulations previously inaccessible to existing numerical schemes. The versatility and accuracy of this code are demonstrated through applications including superradiance, radiative energy transfer, vibrational strong coupling in Bragg resonators, and plasmonic heating of molecular gases. By providing a unified, extensible engine, MaxwellLink potentially offers a powerful platform for exploring emerging phenomena across the research fronts of spectroscopy, quantum optics, plasmonics, and polaritonics.

06 Dec 2025
Predictive atomistic simulations have propelled materials discovery, yet routine setup and debugging still demand computer specialists. This know-how gap limits Integrated Computational Materials Engineering (ICME), where state-of-the-art codes exist but remain cumbersome for non-experts. We address this bottleneck with GENIUS, an AI-agentic workflow that fuses a smart Quantum ESPRESSO knowledge graph with a tiered hierarchy of large language models supervised by a finite-state error-recovery machine. Here we show that GENIUS translates free-form human-generated prompts into validated input files that run to completion on 80% of 295 diverse benchmarks, where 76% are autonomously repaired, with success decaying exponentially to a 7% baseline. Compared with LLM-only baselines, GENIUS halves inference costs and virtually eliminates hallucinations. The framework democratizes electronic-structure DFT simulations by intelligently automating protocol generation, validation, and repair, opening large-scale screening and accelerating ICME design loops across academia and industry worldwide.

05 Dec 2025
Researchers from EPFL's Laboratory of Computational Science and Modeling quantitatively compared the internal latent features of various universal machine-learning interatomic potentials (uMLIPs) using novel reconstruction error metrics. Their analysis revealed distinct ways different uMLIP architectures encode chemical space, the influence of training targets, and the strong pre-training bias retained during fine-tuning.

27 Nov 2025
An algorithm for atom-centered lossy compression of the atomic orbital (AO) basis in density functional theory (DFT) calculations is introduced. This method achieves significant reductions in basis set size, up to 8x for pc-4 basis sets, while maintaining high accuracy, with relative energy errors often below 0.1 kcal/mol for a compression threshold of 10⁻⁵.

25 Nov 2025
Machine-learned interatomic potentials (MLIPs) promise to significantly advance atomistic simulations by delivering quantum-level accuracy for large molecular systems at a fraction of the computational cost of traditional electronic structure methods. While model hubs and categorisation efforts have emerged in recent years, it remains difficult to consistently discover, compare, and apply these models across diverse scenarios. The field still lacks a standardised and comprehensive framework for evaluating MLIP performance. We introduce MLIPAudit, an open, curated and modular benchmarking suite designed to assess the accuracy of MLIP models across a variety of application tasks. MLIPAudit offers a diverse collection of benchmark systems, including small organic compounds, molecular liquids, proteins and flexible peptides, along with pre-computed results for a range of pre-trained and published models. MLIPAudit also provides tools for users to evaluate their models using the same standardised pipeline. A continuously updated leaderboard tracks performance across benchmarks, enabling direct comparison on downstream tasks. By providing a unified, transparent reference framework for model validation and comparison, MLIPAudit aims to foster reproducibility, transparency, and community-driven progress in the development of MLIPs for complex molecular systems. In order to illustrate the use of the library, we present some benchmarks run on a series of internal models, along with publicly available ones (UMA-Small, MACE-OFF, MACE-MP). The library is available on GitHub at this https URL, on PyPI at this https URL under the Apache License 2.0, and the leaderboard can be accessed on HuggingFace at this https URL.

18 Nov 2025
A new algorithm, Trimmed Configuration Interaction (TrimCI), determines quantum many-body ground states without prior knowledge, achieving state-of-the-art accuracy with orders of magnitude fewer determinants by autonomously identifying the most relevant Hilbert space configurations. It demonstrated up to a 10^5-fold efficiency gain in determinant count for complex molecular systems.

13 Nov 2025


Core-shell electrode particles are a promising morphology control strategy for high-performance lithium-ion batteries. However, experimental observations reveal that these structures remain prone to mechanical failure, with shell fractures and core-shell debonding occurring after a single charge. In this work, we present a novel, comprehensive computational framework to predict and gain insight into the failure of core-shell morphologies and the associated degradation in battery performance. The fully coupled chemo-mechano-damage model presented captures the interplay between mechanical damage and electrochemical behaviours, enabling the quantification of particle cracking and capacity fade. Both bulk material fracture and interface debonding are captured by utilising the phase field method. We quantify the severity of particle cracking and capacity loss through case studies on a representative core-shell system (NMC811@NMC532). The results bring valuable insights into cracking patterns, underlying mechanisms, and their impact on capacity loss. Surface cracks are found to initiate when a significantly higher lithium concentration accumulates in the core compared to the shell. Interfacial debonding is shown to arise from localised hoop stresses near the core-shell interface, due to greater shell expansion. This debonding develops rapidly, impedes lithium-ion transport, and can lead to more than 10\% capacity loss after a single discharge. Furthermore, larger particles may experience crack branching driven by extensive tensile zones, potentially fragmenting the entire particle. The framework developed can not only bring new insight into the degradation mechanisms of core-shell particles but also be used to design electrode materials with improved performance and extended lifetime.

05 Nov 2025
Researchers at EPFL developed MatInvent, a reinforcement learning framework that optimizes pre-trained generative diffusion models for inverse inorganic materials design. This approach efficiently generates novel crystal structures with desired properties, achieving up to a 378-fold reduction in expensive property calculations while outperforming conditional generation methods in targeted material discovery.

29 Oct 2025
A unified machine learning interatomic potential (MLIP) achieves ab initio accuracy across molecular, surface, and inorganic crystal systems. The enhanced MACE architecture, coupled with a multi-head replay post-training protocol, reaches state-of-the-art performance across diverse benchmarks, including superior phonon predictions and significant gains in molecular and surface adsorption energies, while demonstrating robust physical realism.
There are no more papers matching your filters at the moment.
