
04 Mar 2025
Researchers at Xanadu developed a method to train generative quantum machine learning models classically, enabling the exploration of quantum circuits with up to 1000 qubits and hundreds of thousands of parameters. This approach demonstrated competitive performance against classical generative models on diverse datasets, highlighting the critical role of quantum coherence for learning.

25 Sep 2021
We propose a teleportation-based scheme to implement a universal set of
quantum gates with a four-component cat code, assisted by appropriate entangled
resource states and photon number resolving detection. The four-component cat
code features the ability to recover from single photon loss. Here, we propose
a concrete procedure to correct the single photon loss, including detecting the
single photon loss event and recovering the initial states. By concatenating
with standard qubit error correcting codes, we estimate the loss threshold for
fault-tolerant quantum computation and obtain a significant improvement over
the two-component cat code.

19 Sep 2025
The discovery of the backpropagation algorithm ranks among one of the most important moments in the history of machine learning, and has made possible the training of large-scale neural networks through its ability to compute gradients at roughly the same computational cost as model evaluation. Despite its importance, a similar backpropagation-like scaling for gradient evaluation of parameterised quantum circuits has remained elusive. Currently, the most popular method requires sampling from a number of circuits that scales with the number of circuit parameters, making training of large-scale quantum circuits prohibitively expensive in practice. Here we address this problem by introducing a class of structured circuits that are not known to be classically simulable and admit gradient estimation with significantly fewer circuits. In the simplest case -- for which the parameters feed into commuting quantum gates -- these circuits allow for fast estimation of the gradient, higher order partial derivatives and the Fisher information matrix. Moreover, specific families of parameterised circuits exist for which the scaling of gradient estimation is in line with classical backpropagation, and can thus be trained at scale. In a toy classification problem on 16 qubits, such circuits show competitive performance with other methods, while reducing the training cost by about two orders of magnitude.
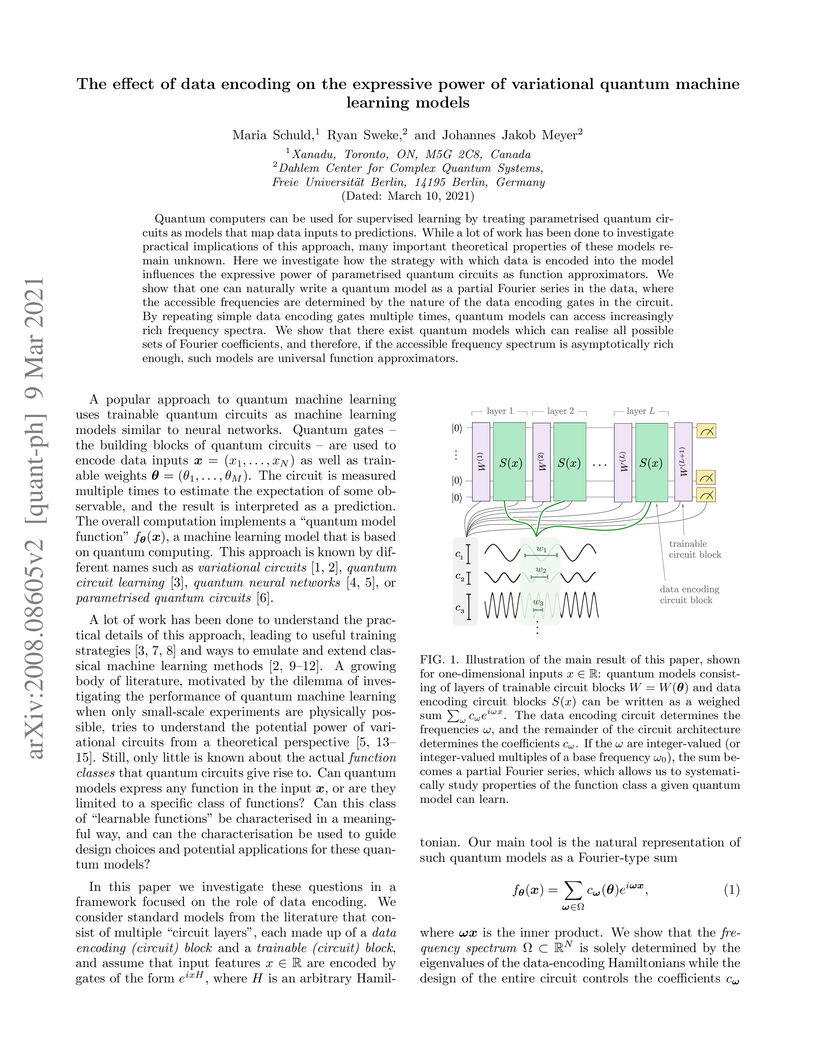
09 Mar 2021
Variational quantum machine learning models with Hamiltonian-based data encoding can be expressed as partial Fourier series. The accessible frequency spectrum is solely determined by the data-encoding Hamiltonians' eigenvalues, while trainable parameters control the Fourier coefficients, allowing repeated encodings to linearly extend the spectrum and proving asymptotic universality for square-integrable functions under ideal conditions.

29 Jul 2022
PennyLane is a Python 3 software framework for differentiable programming of quantum computers. The library provides a unified architecture for near-term quantum computing devices, supporting both qubit and continuous-variable paradigms. PennyLane's core feature is the ability to compute gradients of variational quantum circuits in a way that is compatible with classical techniques such as backpropagation. PennyLane thus extends the automatic differentiation algorithms common in optimization and machine learning to include quantum and hybrid computations. A plugin system makes the framework compatible with any gate-based quantum simulator or hardware. We provide plugins for hardware providers including the Xanadu Cloud, Amazon Braket, and IBM Quantum, allowing PennyLane optimizations to be run on publicly accessible quantum devices. On the classical front, PennyLane interfaces with accelerated machine learning libraries such as TensorFlow, PyTorch, JAX, and Autograd. PennyLane can be used for the optimization of variational quantum eigensolvers, quantum approximate optimization, quantum machine learning models, and many other applications.

14 May 2020

The paper introduces the Quantum Natural Gradient (QNG), an optimization algorithm that leverages the Fubini-Study metric to guide variational quantum algorithms (VQAs) along the information geometry of quantum state space. Numerical experiments demonstrate that QNG consistently achieves faster convergence in fewer iterations compared to standard gradient descent and Adam optimizers for VQAs, particularly as qubit count and circuit depth increase.

21 Aug 2025
The dynamic structure factor (DSF) is a central quantity for interpreting a vast array of inelastic scattering experiments in chemistry and materials science, but its accurate simulation is a considerable challenge for classical computational methods. In this work, we present a quantum algorithm and an end-to-end simulation framework to compute the DSF, providing a general approach for simulating momentum-resolved spectroscopies. We apply this approach to the simulation of electron energy loss spectroscopy (EELS) in the core-level electronic excitation regime, a spectroscopic technique offering sub-nanometer spatial resolution and capable of resolving element-specific information, crucial for analyzing battery materials. We derive a quantum algorithm for computing the DSF for EELS by evaluating the off-diagonal terms of the time-domain Green's function, enabling the simulation of momentum-resolved spectroscopies. To showcase the algorithm, we study the oxygen K-edge EELS spectrum of lithium manganese oxide (), a prototypical cathode material for investigating the mechanisms of oxygen redox in battery materials. For a representative model of an oxygen-centered cluster of with an active space of 18 active orbitals, the algorithm requires a circuit depth of T gates, 100 logical qubits, and roughly shots.

22 Mar 2022
Variational quantum algorithms are ubiquitous in applications of noisy intermediate-scale quantum computers. Due to the structure of conventional parametrized quantum gates, the evaluated functions typically are finite Fourier series of the input parameters. In this work, we use this fact to derive new, general parameter-shift rules for single-parameter gates, and provide closed-form expressions to apply them. These rules are then extended to multi-parameter quantum gates by combining them with the stochastic parameter-shift rule. We perform a systematic analysis of quantum resource requirements for each rule, and show that a reduction in resources is possible for higher-order derivatives. Using the example of the quantum approximate optimization algorithm, we show that the generalized parameter-shift rule can reduce the number of circuit evaluations significantly when computing derivatives with respect to parameters that feed into many gates. Our approach additionally reproduces reconstructions of the evaluated function up to a chosen order, leading to known generalizations of the Rotosolve optimizer and new extensions of the quantum analytic descent optimization algorithm.

03 Mar 2024


Machine learning has emerged recently as a powerful tool for predicting properties of quantum many-body systems. For many ground states of gapped Hamiltonians, generative models can learn from measurements of a single quantum state to reconstruct the state accurately enough to predict local observables. Alternatively, classification and regression models can predict local observables by learning from measurements on different but related states. In this work, we combine the benefits of both approaches and propose the use of conditional generative models to simultaneously represent a family of states, learning shared structures of different quantum states from measurements. The trained model enables us to predict arbitrary local properties of ground states, even for states not included in the training data, without necessitating further training for new observables. We first numerically validate our approach on 2D random Heisenberg models using simulations of up to 45 qubits. Furthermore, we conduct quantum simulations on a neutral-atom quantum computer and demonstrate that our method can accurately predict the quantum phases of square lattices of 1313 Rydberg atoms.

02 Feb 2021





Photonics is the platform of choice to build a modular, easy-to-network quantum computer operating at room temperature. However, no concrete architecture has been presented so far that exploits both the advantages of qubits encoded into states of light and the modern tools for their generation. Here we propose such a design for a scalable and fault-tolerant photonic quantum computer informed by the latest developments in theory and technology. Central to our architecture is the generation and manipulation of three-dimensional hybrid resource states comprising both bosonic qubits and squeezed vacuum states. The proposal enables exploiting state-of-the-art procedures for the non-deterministic generation of bosonic qubits combined with the strengths of continuous-variable quantum computation, namely the implementation of Clifford gates using easy-to-generate squeezed states. Moreover, the architecture is based on two-dimensional integrated photonic chips used to produce a qubit cluster state in one temporal and two spatial dimensions. By reducing the experimental challenges as compared to existing architectures and by enabling room-temperature quantum computation, our design opens the door to scalable fabrication and operation, which may allow photonics to leap-frog other platforms on the path to a quantum computer with millions of qubits.

20 Aug 2025
X-ray absorption spectroscopy (XAS) is a leading technique for understanding structural changes in advanced battery materials such as lithium-excess cathodes. However, extracting critical information like oxidation states from the experimental spectra requires expensive and time-consuming simulations. Building upon a recent proposal to simulate XAS using quantum computers, this work proposes a highly-optimized implementation of the time-domain algorithm for X-ray absorption. Among a host of improvements to Hamiltonian representation, circuit implementation, and measurement strategies, three optimizations are key to the efficiency of the algorithm. The first is the use of product formulas with the compressed double factorized form of the Hamiltonian. The second is recognizing that for spectroscopy applications, it is sufficient to control the error in the eigenvalues of the (approximate) Hamiltonian being implemented by the product formula, rather than the generic error on the full time evolution operator. Using perturbation theory to estimate this eigenvalue error, we find that significantly fewer Trotter steps are needed than expected from the time evolution error bound. The third is the choice of an optimized distribution of samples that takes advantage of the exponentially decaying Lorentzian kernel. Through constant factor resource estimates, we show that a challenging model LiMnO cluster system with 18 spatial orbitals and 22 electrons in the active space can be simulated with 100 logical qubits and less than T gates per circuit. Finally, the algorithm is implemented on a simulator, and the reconstructed spectrum is verified against a classical computational reference. The low cost of our algorithm makes it attractive to use on fault-tolerant quantum devices to accelerate the development and commercialization of high-capacity battery cathodes.

14 Jan 2024
The generation of a logical qubit called the Gottesman-Kitaev-Preskill qubit
in an optical traveling wave is a major challenge for realizing large-scale
universal fault-tolerant optical quantum computers. Recently, probabilistic
generation of elementary GKP qubits has been demonstrated using photon number
measurements and homodyne measurements. However, the generation rate is only a
few Hz, and it will be difficult to generate fault-tolerant GKP qubits at a
practical rate unless success probability is significantly improved. Here, we
propose a method to efficiently synthesize GKP qubits from several quantum
states by adaptive Gaussian operations. In the initial state preparation that
utilizes photon number measurements, an adaptive operation allows any
measurement outcome above a certain threshold to be considered as a success.
This threshold is lowered by utilizing the generalized photon subtraction
method. The initial states are synthesized into a GKP qubit by homodyne
measurements and a subsequent adaptive operation. As a result, the single-shot
success probability of generating fault-tolerant GKP qubits in a realistic
scale system exceeds 10, which is one million times better than previous
methods. This proposal will become a powerful tool for advancing optical
quantum computers from the proof-of-principle stage to practical application.

04 Dec 2025
Efficient encoding of classical data into quantum circuits is a critical challenge that directly impacts the scalability of quantum algorithms. In this work, we present an automated compilation framework for resource-aware quantum data loading tailored to a given input vector and target error tolerance. By explicitly exploiting the trade-off between exact and approximate state preparation, our approach systematically partitions the total error budget between precision and approximation errors, thereby minimizing quantum resource costs. The framework supports a comprehensive suite of state-of-the-art methods, including multiplexer-based loaders, quantum read-only memory (QROM) constructions, sparse encodings, matrix product states (MPS), Fourier series loaders (FSL), and Walsh transform-based diagonal operators. We demonstrate the effectiveness of our framework across several applications, where it consistently uncovers non-obvious, resource-efficient strategies enabled by controlled approximation. In particular, we analyze a computational fluid dynamics workflow where the automated selection of MPS state preparation and Walsh transform-based encoding, combined with a novel Walsh-based measurement technique, leads to resource reductions of over four orders of magnitude compared to previous approaches. We also introduce two independent advances developed through the framework: a more efficient circuit for d-diagonal matrices, and an optimized block encoding for kinetic energy operators. Our results underscore the indispensable role of automated, approximation-aware compilation in making large-scale quantum algorithms feasible on resource-constrained hardware.

17 Apr 2021
With near-term quantum devices available and the race for fault-tolerant quantum computers in full swing, researchers became interested in the question of what happens if we replace a supervised machine learning model with a quantum circuit. While such "quantum models" are sometimes called "quantum neural networks", it has been repeatedly noted that their mathematical structure is actually much more closely related to kernel methods: they analyse data in high-dimensional Hilbert spaces to which we only have access through inner products revealed by measurements. This technical manuscript summarises and extends the idea of systematically rephrasing supervised quantum models as a kernel method. With this, a lot of near-term and fault-tolerant quantum models can be replaced by a general support vector machine whose kernel computes distances between data-encoding quantum states. Kernel-based training is then guaranteed to find better or equally good quantum models than variational circuit training. Overall, the kernel perspective of quantum machine learning tells us that the way that data is encoded into quantum states is the main ingredient that can potentially set quantum models apart from classical machine learning models.

23 Sep 2023
Much is understood about 1-dimensional spin chains in terms of entanglement properties, physical phases, and integrability. However, the Lie algebraic properties of the Hamiltonians describing these systems remain largely unexplored. In this work, we provide a classification of all Lie algebras generated by translation-invariant 2-local spin chain Hamiltonians, or so-called dynamical Lie algebras. We consider chains with open and periodic boundary conditions and find 17 unique dynamical Lie algebras. Our classification covers some well-known models such as the transverse-field Ising model and the Heisenberg chain, and we also find more exotic classes of Hamiltonians that cannot be identified easily. In addition to the closed and open spin chains, we consider systems with a fully connected topology, which may be relevant for quantum machine learning approaches. We discuss the practical implications of our work in the context of quantum control, variational quantum computing, and the spin chain literature.

08 Jan 2025
IQPopt is a software package designed to optimize large-scale instantaneous quantum polynomial circuits on classical hardware. By exploiting an efficient classical simulation algorithm for expectation value estimation, circuits with thousands of qubits and millions of gates can be optimized, provided the relevant objective function has an efficient description in terms of Pauli-Z type observables. Since sampling from instantaneous quantum polynomial circuits is widely believed to be hard for classical computers, this provides a method to identify powerful circuit instances before deployment and sampling on quantum hardware, where computational advantages may exist. The package leverages automatic differentiation in JAX, can be accelerated with access to hardware accelerators such as graphics processing units, and contains a dedicated module that can be used to train and evaluate quantum generative models via the maximum mean discrepancy.

17 May 2024
X-ray absorption spectroscopy is a crucial experimental technique for
elucidating the mechanisms of structural degradation in battery materials.
However, extracting information from the measured spectrum is challenging
without high-quality simulations. In this work, we propose simulating near-edge
X-ray absorption spectra as a promising application for quantum computing. It
is attractive due to the ultralocal nature of X-ray absorption that
significantly reduces the sizes of problems to be simulated, and because of the
classical hardness of simulating spectra. We describe three quantum algorithms
to compute the X-ray absorption spectrum and provide their asymptotic cost. One
of these is a Monte-Carlo based time-domain algorithm, which is cost-friendly
to early fault-tolerant quantum computers. We then apply the framework to an
industrially relevant example, a CAS(22e,18o) active space for an O-Mn cluster
in a Li-excess battery cathode, showing that practically useful simulations
could be obtained with much fewer qubits and gates than ground-state energy
estimation of the same material.

08 Feb 2023
Machine learning is frequently listed among the most promising applications
for quantum computing. This is in fact a curious choice: Today's machine
learning algorithms are notoriously powerful in practice, but remain
theoretically difficult to study. Quantum computing, in contrast, does not
offer practical benchmarks on realistic scales, and theory is the main tool we
have to judge whether it could become relevant for a problem. In this
perspective we explain why it is so difficult to say something about the
practical power of quantum computers for machine learning with the tools we are
currently using. We argue that these challenges call for a critical debate on
whether quantum advantage and the narrative of 'beating' classical machine
learning should continue to dominate the literature the way it does, and
highlight examples for how other perspectives in existing research provide an
important alternative to the focus on advantage.

06 Oct 2023

Neural quantum states have established themselves as a powerful and versatile family of ansatzes for variational Monte Carlo simulations of quantum many-body systems. Of particular prominence are autoregressive neural quantum states (ANQS), which enjoy the expressibility of deep neural networks, and are equipped with a procedure for fast and unbiased sampling. Yet, the non-selective nature of autoregressive sampling makes incorporating quantum number symmetries challenging. In this work, we develop a general framework to make the autoregressive sampling compliant with an arbitrary number of quantum number symmetries. We showcase its advantages by running electronic structure calculations for a range of molecules with multiple symmetries of this kind. We reach the level of accuracy reported in previous works with more than an order of magnitude speedup and achieve chemical accuracy for all studied molecules, which is a milestone unreported so far. Combined with the existing effort to incorporate space symmetries, our approach expands the symmetry toolbox essential for any variational ansatz and brings the ANQS closer to being a competitive choice for studying challenging quantum many-body systems.

05 Oct 2025
Accurate state preparation is a critical bottleneck in many quantum algorithms, particularly those for ground state energy estimation. Even in fault-tolerant quantum computing, preparing a quantum state with sufficient overlap to the desired eigenstate remains a major challenge. To address this, we develop a unified framework for filtered-state preparation that enhances the overlap of a given input state through spectral filtering. This framework encompasses the polynomial and trigonometric realizations of filters, allowing a transparent analysis of the trade-offs between overlap amplification and preparation cost. As examples, we introduce signal-processing-inspired filters, such as Gaussian filters and Krylov subspace-based filters, that adaptively suppress excited-state contributions using low-rank projections. Within this framework, we further develop a filtered variant of QPE (FQPE) that mitigates the unfavorable dependence on the initial overlap present in standard QPE. Numerical experiments on Fermi-Hubbard models show that FQPE reduces the total runtime by more than two orders of magnitude in the high-precision regime, with overlap amplification exceeding a factor of one hundred.
There are no more papers matching your filters at the moment.
