
27 May 2025
Classical methods to simulate quantum systems are not only a key element of
the physicist's toolkit for studying many-body models but are also increasingly
important for verifying and challenging upcoming quantum computers. Pauli
propagation has recently emerged as a promising new family of classical
algorithms for simulating digital quantum systems. Here we provide a
comprehensive account of Pauli propagation, tracing its algorithmic structure
from its bit-level implementation and formulation as a tree-search problem, all
the way to its high-level user applications for simulating quantum circuits and
dynamics. Utilising these observations, we present PauliPropagation.jl, a Julia
software package that can perform rapid Pauli propagation simulation straight
out-of-the-box and can be used more generally as a building block for novel
simulation algorithms.

29 Mar 2025
We introduce Majorana Propagation, an algorithmic framework for the classical
simulation of Fermionic circuits. Inspired by Pauli Propagation, Majorana
Propagation operates by applying successive truncations throughout the
Heisenberg evolution of the observable. We identify monomial length as an
effective truncation strategy for typical, unstructured circuits by proving
that high-length Majorana monomials are exponentially unlikely to contribute to
expectation values and the backflow of high-length monomials to lower-length
monomials is quadratically suppressed. Majorana Propagation can be used either
independently, or in conjunction with quantum hardware, to simulate Fermionic
systems relevant to quantum chemistry and condensed matter. We exemplify this
by using Majorana Propagation to find circuits that approximate ground states
for strongly correlated systems of up to 52 Fermionic modes. Our results
indicate that Majorana Propagation is orders of magnitude faster and more
accurate than state-of-the-art tensor-network-based circuit simulators.

04 Nov 2025
Accurate estimation of observables in quantum systems is a central challenge in quantum information science, yet practical implementations are fundamentally constrained by the limited number of measurement shots. In this work we explore a variation of the classical shadows protocol in which the measurements are kept local while allowing the resulting classical shadows themselves to be correlated. By constructing locally optimal shadows, we obtain unbiased estimators that outperform state-of-the-art methods, achieving the same accuracy with substantially fewer measurements. We validate our approach through numerical experiments on molecular Hamiltonians with up to 40 qubits and a 50-qubit Ising model consistently observing significant reductions in estimation errors.

10 Jan 2025
 California Institute of Technology
California Institute of Technology University College London
University College London University of OxfordTechnology Innovation InstituteIBM QuantumUniversity of HelsinkiAWS Center for Quantum ComputingTrinity College DublinAlgorithmiq Ltd.Ecole Polytechnique Fédérale de LausannePhasecraft Ltd.HUN-REN Wigner Research Centre for PhysicsQuantum MotionHUN-REN Alfréd Rényi Institute of MathematicsGoogle Quantum AIUniversità di FirenzeICREA (Institucio Catalana de Recerca i Estudis Avançats)ICFO
Institut de Ciencies FotoniquesINFN
Sezione di Firenze
University of OxfordTechnology Innovation InstituteIBM QuantumUniversity of HelsinkiAWS Center for Quantum ComputingTrinity College DublinAlgorithmiq Ltd.Ecole Polytechnique Fédérale de LausannePhasecraft Ltd.HUN-REN Wigner Research Centre for PhysicsQuantum MotionHUN-REN Alfréd Rényi Institute of MathematicsGoogle Quantum AIUniversità di FirenzeICREA (Institucio Catalana de Recerca i Estudis Avançats)ICFO
Institut de Ciencies FotoniquesINFN
Sezione di FirenzeA team of 22 researchers from leading quantum computing companies and academic institutions examines six prevalent 'myths' in quantum computing discourse. The paper offers a balanced perspective on the field's progress, arguing that techniques like Quantum Error Mitigation and Variational Quantum Algorithms will continue to be important as quantum hardware evolves towards fault tolerance.

21 Dec 2022
We present a self consistent field approach (SCF) within the Adaptive Derivative-Assembled Problem-Tailored Ansatz Variational Quantum Eigensolver (ADAPT-VQE) framework for efficient quantum simulations of chemical systems on near-term quantum computers. To this end, our ADAPT-VQE-SCF approach combines the idea of generating an ansatz with a small number of parameters, resulting in shallow-depth quantum circuits with a direct minimization of an energy expression which is correct to second order with respect to changes in the molecular orbital basis. Our numerical analysis, including calculations for the transition metal complex ferrocene (Fe), indicates that convergence in the self-consistent orbital optimization loop can be reached without a considerable increase in the number of two-qubit gates in the quantum circuit by comparison to a VQE optimization in the initial molecular orbital basis. Moreover, the orbital optimization can be carried out simultaneously within each iteration of the ADAPT-VQE cycle. ADAPT-VQE-SCF thus allows us to implement a routine analogous to CASSCF, a cornerstone of state-of-the-art computational chemistry, in a hardware-efficient manner on near-term quantum computers. Hence, ADAPT-VQE-SCF paves the way towards a paradigm shift for quantitative quantum-chemistry simulations on quantum computers by requiring fewer qubits and opening up for the use of large and flexible atomic orbital basis sets in contrast to earlier methods that are predominantly based on the idea of full active spaces with minimal basis sets.
21 Nov 2024
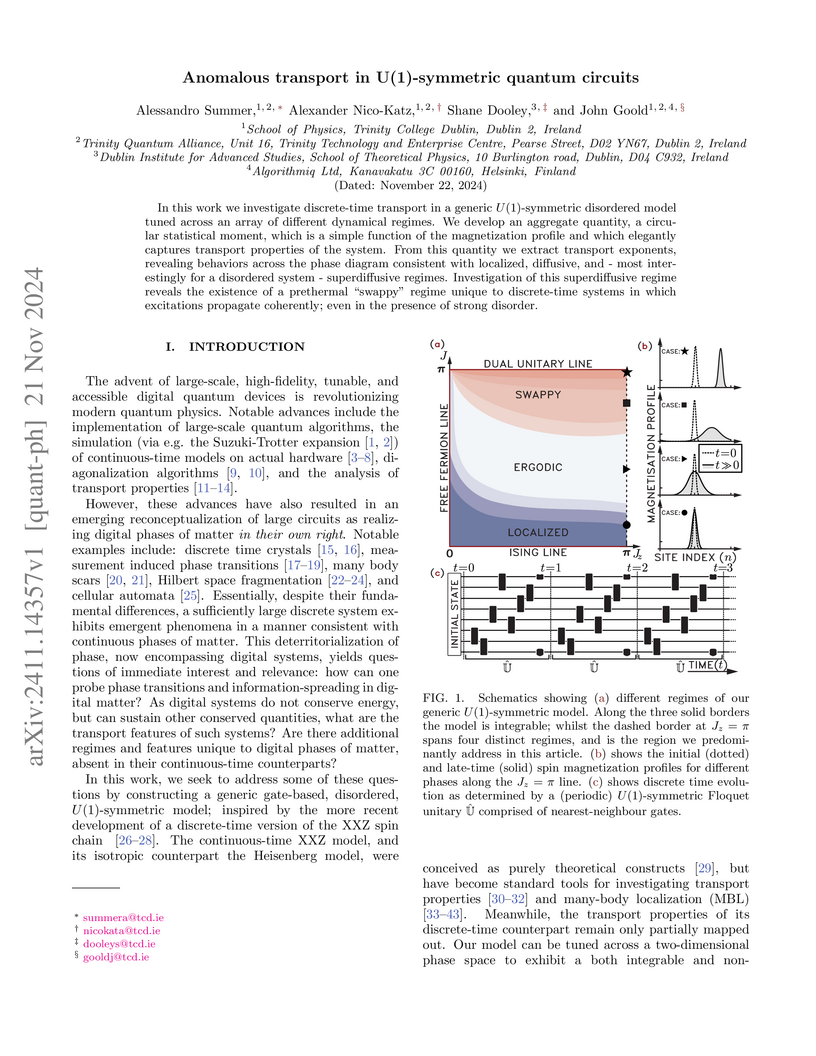
In this work we investigate discrete-time transport in a generic U(1)-symmetric disordered model tuned across an array of different dynamical regimes. We develop an aggregate quantity, a circular statistical moment, which is a simple function of the magnetization profile and which elegantly captures transport properties of the system. From this quantity we extract transport exponents, revealing behaviors across the phase diagram consistent with localized, diffusive, and - most interestingly for a disordered system - superdiffusive regimes. Investigation of this superdiffusive regime reveals the existence of a prethermal "swappy" regime unique to discrete-time systems in which excitations propagate coherently; even in the presence of strong disorder.

24 Dec 2024
Recent studies of organic molecular solids are highlighted by their complex phase diagram and light-induced phenomena, such as Mott insulator, spin liquid phase, and superconductivity. However, a discrepancy between experimental observation and first-principle calculation on the -(BEDT-TTF)X family inhibits understanding their properties. Here, we revisit the electronic structure of -(BEDT-TTF)Cu(CN) with the recently developed DFT+GOU method to correct the energy level of molecular orbital states in the molecular solid. Our work reveals that the insulating electronic structure of -(BEDT-TTF)Cu(CN) originates from the energy gap between the highest occupied and the lowest unoccupied molecular orbital states of the BEDT-TTF dimers, that are the periodic unit of the molecular solid. We verify that our calculation result provides consistent band gap, optical conductivity, and evolution of the metal-insulator transition as a function of pressure with experimental observations. Especially, the superconducting dome of -(BEDT-TTF)Cu(CN), which originates from the flat band state at the Fermi level, is reproduced. Additionally, we constructed a new low-energy lattice model based on the ability of electronic structure data that can be used to address many-body physics, such as quantum spin liquid and double-holon dynamics. Our provides a deeper understanding of the complex phase diagram and various light-induced phenomena in the -(BEDT-TTF)X family and the other complex organic molecular solids.

01 Aug 2023

In recent years, there has been tremendous progress in the development of quantum computing hardware, algorithms and services leading to the expectation that in the near future quantum computers will be capable of performing simulations for natural science applications, operations research, and machine learning at scales mostly inaccessible to classical computers. Whereas the impact of quantum computing has already started to be recognized in fields such as cryptanalysis, natural science simulations, and optimization among others, very little is known about the full potential of quantum computing simulations and machine learning in the realm of healthcare and life science (HCLS). Herein, we discuss the transformational changes we expect from the use of quantum computation for HCLS research, more specifically in the field of cell-centric therapeutics. Moreover, we identify and elaborate open problems in cell engineering, tissue modeling, perturbation modeling, and bio-topology while discussing candidate quantum algorithms for research on these topics and their potential advantages over classical computational approaches.

30 Mar 2025

We introduce the Piquasso quantum programming framework, a full-stack open-source software platform for the simulation and programming of photonic quantum computers. Piquasso can be programmed via a high-level Python programming interface enabling users to perform efficient quantum computing with discrete and continuous variables. Via optional high-performance C++ backends, Piquasso provides state-of-the-art performance in the simulation of photonic quantum computers. The Piquasso framework is supported by an intuitive web-based graphical user interface where the users can design quantum circuits, run computations, and visualize the results.

01 Nov 2024
Quantum circuits with local unitaries have emerged as a rich playground for the exploration of many-body quantum dynamics of discrete-time systems. While the intrinsic locality makes them particularly suited to run on current quantum processors, the task of verification at non-trivial scales is complicated for non-integrable systems. Here, we study a special class of maximally chaotic circuits known as dual unitary circuits -- exhibiting unitarity in both space and time -- that are known to have exact analytical solutions for certain correlation functions. With advances in noise learning and the implementation of novel error mitigation methods, we show that a superconducting quantum processor with 91 qubits is able to accurately simulate these correlators. We then probe dynamics beyond exact verification, by perturbing the circuits away from the dual unitary point, and compare our results to classical approximations with tensor networks. These results cement error-mitigated digital quantum simulation on pre-fault-tolerant quantum processors as a trustworthy platform for the exploration and discovery of novel emergent quantum many-body phases.

24 Jul 2023
Until fault-tolerance becomes implementable at scale, quantum computing will heavily rely on noise mitigation techniques. While methods such as zero noise extrapolation with probabilistic error amplification (ZNE-PEA) and probabilistic error cancellation (PEC) have been successfully tested on hardware recently, their scalability to larger circuits may be limited. Here, we introduce the tensor-network error mitigation (TEM) algorithm, which acts in post-processing to correct the noise-induced errors in estimations of physical observables. The method consists of the construction of a tensor network representing the inverse of the global noise channel affecting the state of the quantum processor, and the consequent application of the map to informationally complete measurement outcomes obtained from the noisy state. TEM does therefore not require additional quantum operations other than the implementation of informationally complete POVMs, which can be achieved through randomised local measurements. The key advantage of TEM is that the measurement overhead is quadratically smaller than in PEC. We test TEM extensively in numerical simulations in different regimes. We find that TEM can be applied to circuits of twice the depth compared to what is achievable with PEC under realistic conditions with sparse Pauli-Lindblad noise, such as those in [E. van den Berg et al., Nat. Phys. (2023)]. By using Clifford circuits, we explore the capabilities of the method in wider and deeper circuits with lower noise levels. We find that in the case of 100 qubits and depth 100, both PEC and ZNE fail to produce accurate results by using shots, while TEM succeeds.

23 Jul 2025
We develop and demonstrate methods for simulating the scattering of particle wave packets in the interacting Thirring model on digital quantum computers, with hardware implementations on up to 80 qubits. We identify low-entanglement time slices of the scattering dynamics and exploit their efficient representation by tensor networks. Circuit compression based on matrix product state techniques yields on average a reduction by a factor of 3.2 in circuit depth compared to conventional approaches, allowing longer evolution times to be evaluated with higher fidelity on contemporary quantum processors. Utilizing zero-noise extrapolation in combination with Pauli twirling, on quantum hardware we accurately simulate the full scattering dynamics on 40 qubits, and further demonstrate the wave packet state-preparation on 80 qubits.

22 Jun 2022

Scientific and technological advances in medicine and systems biology have unequivocally shown that health and disease must be viewed in the context of the interplay among multiple molecular and environmental factors. Understanding the effects of cellular interconnection on disease progression may lead to the identification of novel disease genes and pathways, and hence influence precision diagnostics and therapeutics. To accomplish this goal, the emerging field of network medicine applies network science approaches to investigate disease pathogenesis, integrating information from relevant Omics databases, including protein-protein interaction, correlation-based, gene regulatory, and Bayesian networks. However, this requires analysing and computing large amounts of data. Moreover, if we are to efficiently search for new drugs and new drug combinations, there is a pressing need for computational methods that could allow us to access the immense chemical compound space until now largely unexplored. Finally, at the microscopic level, drug-target chemistry simulation is ultimately a quantum problem, and hence it requires a quantum solution. As we will discuss, quantum computing may be a key ingredient in enabling the full potential of network medicine. We propose to combine network medicine and quantum algorithms in a novel research field, quantum network medicine, to lay the foundations of a new era of disease prevention and drug design.

15 Nov 2022
In this work, we report on a novel quantum gate approximation algorithm based
on the application of parametric two-qubit gates in the synthesis process. The
utilization of these parametric two-qubit gates in the circuit design allows us
to transform the discrete combinatorial problem of circuit synthesis into an
optimization problem over continuous variables. The circuit is then compressed
by a sequential removal of two-qubit gates from the design, while the remaining
building blocks are continuously adapted to the reduced gate structure by
iterated learning cycles. We implemented the developed algorithm in the
SQUANDER software package and benchmarked it against several state-of-the-art
quantum gate synthesis tools. Our numerical experiments revealed outstanding
circuit compression capabilities of our compilation algorithm providing the
most optimal gate count in the majority of the addressed quantum circuits.

09 Dec 2024
Efficient and effective compilation of quantum circuits remains an important
aspect of executing quantum programs. In this paper, we propose a generic
compilation framework particularly suitable for limited connectivity, that
extends many of the known techniques for Pauli network synthesis. Our
compilation method, built on the introduced Clifford Executive Representation,
stands out by considering the implementation of multiple Pauli operators at
once, which are not necessarily commuting. The proposed technique also allows
for effective Clifford circuit synthesis. We benchmark our methods against
circuits resulting from the Variational Quantum Eigensolver algorithm and the
results show that the proposed methods outperform the state-of-the-art.

28 Oct 2023
The amount of work that can be extracted from a quantum system can be increased by exploiting the information obtained from a measurement performed on a correlated ancillary system. The concept of daemonic ergotropy has been introduced to properly describe and quantify this work extraction enhancement in the quantum regime. We here explore the application of this idea in the context of continuously-monitored open quantum systems, where information is gained by measuring the environment interacting with the energy-storing quantum device. We first show that the corresponding daemonic ergotropy takes values between the ergotropy and the energy of the corresponding unconditional state. The upper bound is achieved by assuming an initial pure state and a perfectly efficient projective measurement on the environment, independently of the kind of measurement performed. On the other hand, if the measurement is inefficient or the initial state is mixed, the daemonic ergotropy is generally dependent on the measurement strategy. This scenario is investigated via a paradigmatic example of an open quantum battery: a two-level atom driven by a classical field and whose spontaneously emitted photons are continuously monitored via either homodyne, heterodyne, or photo-detection.

13 Jul 2025
Decomposing unitary operations into native gates is an essential step for implementing quantum algorithms. For qubit-based devices, where native gates are typically single- and two-qubit operations, a range of decomposition techniques have been developed. In particular, efficient algorithms exist for decomposing exponentials of Pauli strings while taking hardware topology in account. Motivated by the growing interest in qutrit-based quantum computing, we develop analogous decomposition methods for qutrit systems. Specifically, we introduce an algorithm that decomposes the exponential of an arbitrary tensor product of Weyl-Heisenberg operators (plus their Hermitian conjugation) into single- and two-qutrit gates. We further extend this approach to unitaries generated by Gell-Mann string (i.e., a tensor product of Gell-Mann matrices). Since both Gell-Mann matrices and Weyl-Heisenberg operators form (together with identity) complete operator bases of qutrit operators, we can use this result also to decompose any multi-qutrit gate that is diagonal up to single-qutrit rotations. As a practical application, we use our method to decompose the layers of the quantum approximate optimization algorithm for qutrit-based implementations of the graph k-coloring problem. For values of well-suited to qutrit architectures (e.g., or in general ), our approach yields significantly shallower circuits compared to qubit-based implementations, an advantage that grows with problem size, while also requiring a smaller total Hilbert space dimension. Finally, we also address the routing challenge in qutrit architectures that arises due to the limited connectivity of the devices. In particular, we generalize the Steiner-Gauss method, originally developed to reduce CNOT counts in qubit circuit, to optimize gate routing in qutrit-based systems.

29 May 2024
The understanding of open quantum systems is crucial for the development of
quantum technologies. Of particular relevance is the characterisation of
divisible quantum dynamics, seen as a generalisation of Markovian processes to
the quantum setting. Here, we propose a way to detect divisibility and quantify
how non-divisible a quantum channel is through the concept of channel
discrimination. We ask how well we can distinguish generic dynamics from
divisible dynamics. We show that this question can be answered efficiently
through semidefinite programming, which provides us with an operational and
efficient way to quantify divisibility.

20 Feb 2024


We formulate and implement the Variational Quantum Eigensolver Self Consistent Field (VQE-SCF) algorithm in combination with polarizable embedding (PE), thereby extending PE to the regime of quantum computing. We test the resulting algorithm, PE-VQE-SCF, on quantum simulators and demonstrate that the computational stress on the quantum device is only slightly increased in terms of gate counts compared to regular VQE-SCF. On the other hand, no increase in shot noise was observed. We illustrate how PE-VQE-SCF may lead to the modeling of real chemical systems using a simulation of the reaction barrier of the Diels-Alder reaction between furan and ethene as an example.

11 Jul 2025


Proton transfer reactions are fundamental to many chemical and biological systems, where quantum effects such as tunneling, delocalization, and zero-point motion play key kinetic control roles. However, classical methods capable of accurately capturing these phenomena scale prohibitively with system size. Here, we develop and demonstrate quantum computing algorithms based on the Nuclear-Electronic Orbital framework, treating the transferring proton quantum mechanically. We assess the potential of current quantum devices for simulating proton transfer kinetics with high accuracy. We first construct a deep initial ansätze within a truncated orbital space by employing the frozen natural orbital approximation. Then, to balance circuit depth against state fidelity, we implement an adaptive form of approximate quantum compiling. Using resulting circuits at varying compression levels transpiled for the ibm_fez device, we compute barrier heights and delocalised proton densities along the proton transfer pathway using a realistic hardware noise model. We find that, although current quantum hardware introduces significant noise relative to the demanding energy tolerances involved, our approach allows substantial circuit simplification while maintaining energy barrier estimates within 13% of the reference value. Despite present hardware limitations, these results offer a practical means of approximating key circuit segments in near-term devices and early fault-tolerant quantum computing systems.
There are no more papers matching your filters at the moment.
