
09 Dec 2025
Enzymes are crucial catalysts that enable a wide range of biochemical reactions. Efficiently identifying specific enzymes from vast protein libraries is essential for advancing biocatalysis. Traditional computational methods for enzyme screening and retrieval are time-consuming and resource-intensive. Recently, deep learning approaches have shown promise. However, these methods focus solely on the interaction between enzymes and reactions, overlooking the inherent hierarchical relationships within each domain. To address these limitations, we introduce FGW-CLIP, a novel contrastive learning framework based on optimizing the fused Gromov-Wasserstein distance. FGW-CLIP incorporates multiple alignments, including inter-domain alignment between reactions and enzymes and intra-domain alignment within enzymes and reactions. By introducing a tailored regularization term, our method minimizes the Gromov-Wasserstein distance between enzyme and reaction spaces, which enhances information integration across these domains. Extensive evaluations demonstrate the superiority of FGW-CLIP in challenging enzyme-reaction tasks. On the widely-used EnzymeMap benchmark, FGW-CLIP achieves state-of-the-art performance in enzyme virtual screening, as measured by BEDROC and EF metrics. Moreover, FGW-CLIP consistently outperforms across all three splits of ReactZyme, the largest enzyme-reaction benchmark, demonstrating robust generalization to novel enzymes and reactions. These results position FGW-CLIP as a promising framework for enzyme discovery in complex biochemical settings, with strong adaptability across diverse screening scenarios.

02 Dec 2025
Accurately predicting protein fitness with minimal experimental data is a persistent challenge in protein engineering. We introduce PRIMO (PRotein In-context Mutation Oracle), a transformer-based framework that leverages in-context learning and test-time training to adapt rapidly to new proteins and assays without large task-specific datasets. By encoding sequence information, auxiliary zero-shot predictions, and sparse experimental labels from many assays as a unified token set in a pre-training masked-language modeling paradigm, PRIMO learns to prioritize promising variants through a preference-based loss function. Across diverse protein families and properties-including both substitution and indel mutations-PRIMO outperforms zero-shot and fully supervised baselines. This work underscores the power of combining large-scale pre-training with efficient test-time adaptation to tackle challenging protein design tasks where data collection is expensive and label availability is limited.

28 Nov 2025
Protein inverse folding, the design of an amino acid sequence based on a target 3D structure, is a fundamental problem of computational protein engineering. Existing methods either generate sequences without leveraging external knowledge or relying on protein language models (PLMs). The former omits the evolutionary information stored in protein databases, while the latter is parameter-inefficient and inflexible to adapt to ever-growing protein data. To overcome the above drawbacks, in this paper we propose a novel method, called retrieval-augmented denoising diffusion (RadDiff), for protein inverse folding. Given the target protein backbone, RadDiff uses a hierarchical search strategy to efficiently retrieve structurally similar proteins from large protein databases. The retrieved structures are then aligned residue-by-residue to the target to construct a position-specific amino acid profile, which serves as an evolutionary-informed prior that conditions the denoising process. A lightweight integration module is further designed to incorporate this prior effectively. Experimental results on the CATH, PDB, and TS50 datasets show that RadDiff consistently outperforms existing methods, improving sequence recovery rate by up to 19%. Experimental results also demonstrate that RadDiff generates highly foldable sequences and scales effectively with database size.

03 Dec 2025
The RNA inverse folding problem, a key challenge in RNA design, involves identifying nucleotide sequences that can fold into desired secondary structures, which are critical for ensuring molecular stability and function. The inherent complexity of this task stems from the intricate relationship between sequence and structure, making it particularly challenging. In this paper, we propose a framework, named HyperRNA, a generative model with an encoder-decoder architecture that leverages hypergraphs to design RNA sequences. Specifically, our HyperRNA model consists of three main components: preprocessing, encoding and decoding.
In the preprocessing stage, graph structures are constructed by extracting the atom coordinates of RNA backbone based on 3-bead coarse-grained representation. The encoding stage processes these graphs, capturing higher order dependencies and complex biomolecular interactions using an attention embedding module and a hypergraph-based encoder. Finally, the decoding stage generates the RNA sequence in an autoregressive manner. We conducted quantitative and qualitative experiments on the PDBBind and RNAsolo datasets to evaluate the inverse folding task for RNA sequence generation and RNA-protein complex sequence generation. The experimental results demonstrate that HyperRNA not only outperforms existing RNA design methods but also highlights the potential of leveraging hypergraphs in RNA engineering.

29 Nov 2025
VCWorld, a biological world model developed by researchers at Shanghai Jiao Tong University and NeoLife AI, simulates cellular responses to drug perturbations by integrating an open-world biological knowledge graph with large language model-driven reasoning. This model provides interpretable, stepwise predictions and mechanistic hypotheses, achieving state-of-the-art predictive performance on a new gene-centric benchmark while enhancing data efficiency.

02 Dec 2025
Wastewater-based genomic surveillance has emerged as a powerful tool for population-level viral monitoring, offering comprehensive insights into circulating viral variants across entire communities. However, this approach faces significant computational challenges stemming from high sequencing noise, low viral coverage, fragmented reads, and the complete absence of labeled variant annotations. Traditional reference-based variant calling pipelines struggle with novel mutations and require extensive computational resources. We present a comprehensive framework for unsupervised viral variant detection using Vector-Quantized Variational Autoencoders (VQ-VAE) that learns discrete codebooks of genomic patterns from k-mer tokenized sequences without requiring reference genomes or variant labels. Our approach extends the base VQ-VAE architecture with masked reconstruction pretraining for robustness to missing data and contrastive learning for highly discriminative embeddings. Evaluated on SARS-CoV-2 wastewater sequencing data comprising approximately 100,000 reads, our VQ-VAE achieves 99.52% mean token-level accuracy and 56.33% exact sequence match rate while maintaining 19.73% codebook utilization (101 of 512 codes active), demonstrating efficient discrete representation learning. Contrastive fine-tuning with different projection dimensions yields substantial clustering improvements: 64-dimensional embeddings achieve +35% Silhouette score improvement (0.31 to 0.42), while 128-dimensional embeddings achieve +42% improvement (0.31 to 0.44), clearly demonstrating the impact of embedding dimensionality on variant discrimination capability. Our reference-free framework provides a scalable, interpretable approach to genomic surveillance with direct applications to public health monitoring.

02 Dec 2025
Single-cell RNA sequencing (scRNA-seq) is essential for decoding tumor heterogeneity. However, pan-cancer research still faces two key challenges: learning discriminative and efficient single-cell representations, and establishing a comprehensive evaluation benchmark. In this paper, we introduce PanFoMa, a lightweight hybrid neural network that combines the strengths of Transformers and state-space models to achieve a balance between performance and efficiency. PanFoMa consists of a front-end local-context encoder with shared self-attention layers to capture complex, order-independent gene interactions; and a back-end global sequential feature decoder that efficiently integrates global context using a linear-time state-space model. This modular design preserves the expressive power of Transformers while leveraging the scalability of Mamba to enable transcriptome modeling, effectively capturing both local and global regulatory signals. To enable robust evaluation, we also construct a large-scale pan-cancer single-cell benchmark, PanFoMaBench, containing over 3.5 million high-quality cells across 33 cancer subtypes, curated through a rigorous preprocessing pipeline. Experimental results show that PanFoMa outperforms state-of-the-art models on our pan-cancer benchmark (+4.0\%) and across multiple public tasks, including cell type annotation (+7.4\%), batch integration (+4.0\%) and multi-omics integration (+3.1\%). The code is available at this https URL.

26 Nov 2025
Researchers at Tencent AI for Life Sciences Lab introduce Balanced Fine-Tuning (BFT), an efficient post-training method to align Large Language Models with biomedical knowledge. BFT consistently surpasses Supervised Fine-Tuning across various biomedical tasks, mitigates catastrophic forgetting, and enables LLMs to generate semantically rich gene and cell embeddings for downstream applications like single-cell analysis and perturbation response prediction.
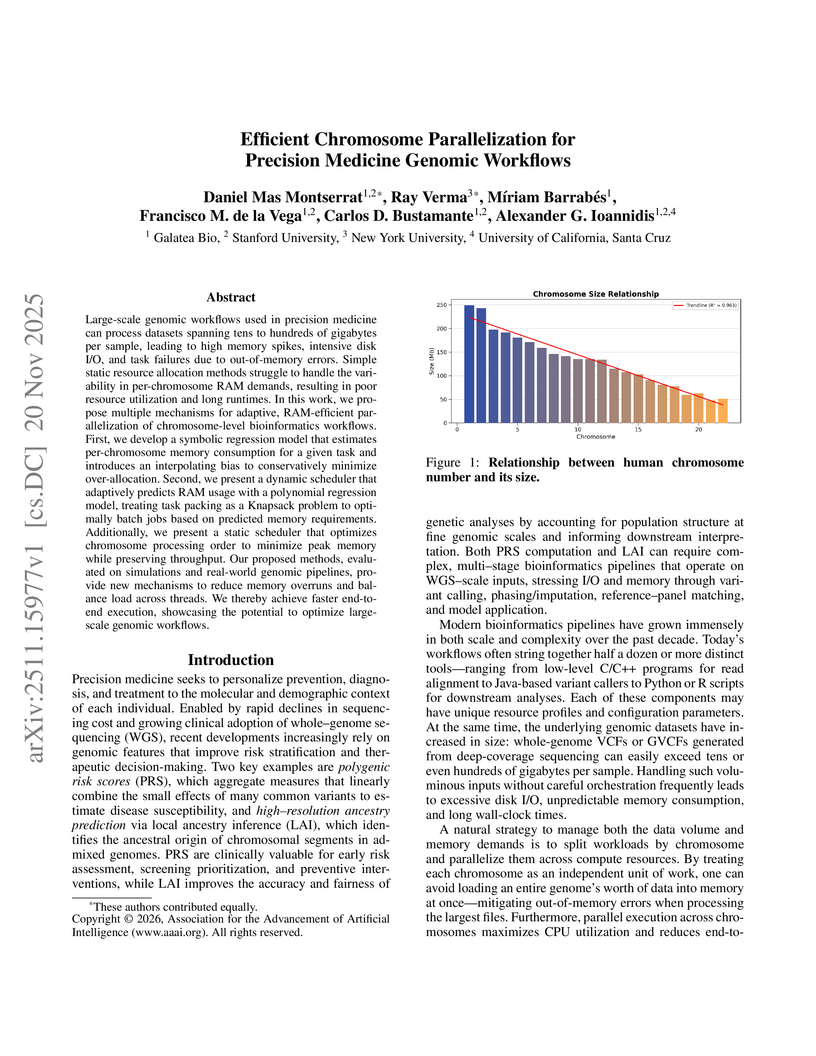
20 Nov 2025
Large-scale genomic workflows used in precision medicine can process datasets spanning tens to hundreds of gigabytes per sample, leading to high memory spikes, intensive disk I/O, and task failures due to out-of-memory errors. Simple static resource allocation methods struggle to handle the variability in per-chromosome RAM demands, resulting in poor resource utilization and long runtimes. In this work, we propose multiple mechanisms for adaptive, RAM-efficient parallelization of chromosome-level bioinformatics workflows. First, we develop a symbolic regression model that estimates per-chromosome memory consumption for a given task and introduces an interpolating bias to conservatively minimize over-allocation. Second, we present a dynamic scheduler that adaptively predicts RAM usage with a polynomial regression model, treating task packing as a Knapsack problem to optimally batch jobs based on predicted memory requirements. Additionally, we present a static scheduler that optimizes chromosome processing order to minimize peak memory while preserving throughput. Our proposed methods, evaluated on simulations and real-world genomic pipelines, provide new mechanisms to reduce memory overruns and balance load across threads. We thereby achieve faster end-to-end execution, showcasing the potential to optimize large-scale genomic workflows.

19 Nov 2025

Recent advances in computational pathology have leveraged vision-language models to learn joint representations of Hematoxylin and Eosin (HE) images with spatial transcriptomic (ST) profiles. However, existing approaches typically align HE tiles with their corresponding ST profiles at a single scale, overlooking fine-grained cellular structures and their spatial organization. To address this, we propose Sigmma, a multi-modal contrastive alignment framework for learning hierarchical representations of HE images and spatial transcriptome profiles across multiple scales. Sigmma introduces multi-scale contrastive alignment, ensuring that representations learned at different scales remain coherent across modalities. Furthermore, by representing cell interactions as a graph and integrating inter- and intra-subgraph relationships, our approach effectively captures cell-cell interactions, ranging from fine to coarse, within the tissue microenvironment. We demonstrate that Sigmm learns representations that better capture cross-modal correspondences, leading to an improvement of avg. 9.78\% in the gene-expression prediction task and avg. 26.93\% in the cross-modal retrieval task across datasets. We further show that it learns meaningful multi-tissue organization in downstream analyses.

19 Nov 2025
Researchers from Wuhan University and Tsinghua University developed TDAM-CRC, a deep learning model that significantly improves prognostic stratification for colorectal cancer patients using whole-slide images, achieving a C-index of 0.747 on the TCGA cohort and outperforming existing methods. The study also uncovered molecular drivers, an immunosuppressive tumor microenvironment in high-risk tumors, and identified MRPL37 as an independent favorable prognostic biomarker, integrated into a robust clinical nomogram.

20 Nov 2025
Open-weight bio-foundation models present a dual-use dilemma. While holding great promise for accelerating scientific research and drug development, they could also enable bad actors to develop more deadly bioweapons. To mitigate the risk posed by these models, current approaches focus on filtering biohazardous data during pre-training. However, the effectiveness of such an approach remains unclear, particularly against determined actors who might fine-tune these models for malicious use. To address this gap, we propose BioRiskEval, a framework to evaluate the robustness of procedures that are intended to reduce the dual-use capabilities of bio-foundation models. BioRiskEval assesses models' virus understanding through three lenses, including sequence modeling, mutational effects prediction, and virulence prediction. Our results show that current filtering practices may not be particularly effective: Excluded knowledge can be rapidly recovered in some cases via fine-tuning, and exhibits broader generalizability in sequence modeling. Furthermore, dual-use signals may already reside in the pretrained representations, and can be elicited via simple linear probing. These findings highlight the challenges of data filtering as a standalone procedure, underscoring the need for further research into robust safety and security strategies for open-weight bio-foundation models.

22 Oct 2025
Researchers from the Chinese Academy of Sciences introduce KnowMol, a Molecular Large Language Model that enhances molecular comprehension through a multi-level chemical knowledge dataset (KnowMol-100K) and specialized representation strategies for 1D strings (SELFIES with dedicated vocabulary) and 2D graphs (hierarchical encoder). KnowMol achieves superior performance across molecular understanding and generation tasks, evidenced by a BLEU-4 score of 0.595 in molecule captioning and an Exact Match of 0.752 in forward reaction prediction, while operating with reduced pre-training computational costs.

30 Oct 2025




Scientific Large Language Models (Sci-LLMs) have emerged as a promising frontier for accelerating biological discovery. However, these models face a fundamental challenge when processing raw biomolecular sequences: the tokenization dilemma. Whether treating sequences as a specialized language, risking the loss of functional motif information, or as a separate modality, introducing formidable alignment challenges, current strategies fundamentally limit their reasoning capacity. We challenge this sequence-centric paradigm by positing that a more effective strategy is to provide Sci-LLMs with high-level structured context derived from established bioinformatics tools, thereby bypassing the need to interpret low-level noisy sequence data directly. Through a systematic comparison of leading Sci-LLMs on biological reasoning tasks, we tested three input modes: sequence-only, context-only, and a combination of both. Our findings are striking: the context-only approach consistently and substantially outperforms all other modes. Even more revealing, the inclusion of the raw sequence alongside its high-level context consistently degrades performance, indicating that raw sequences act as informational noise, even for models with specialized tokenization schemes. These results suggest that the primary strength of existing Sci-LLMs lies not in their nascent ability to interpret biomolecular syntax from scratch, but in their profound capacity for reasoning over structured, human-readable knowledge. Therefore, we argue for reframing Sci-LLMs not as sequence decoders, but as powerful reasoning engines over expert knowledge. This work lays the foundation for a new class of hybrid scientific AI agents, repositioning the developmental focus from direct sequence interpretation towards high-level knowledge synthesis. The code is available at this https URL.

14 Nov 2025



This paper introduces an innovative Electronic Health Record (EHR) foundation model that integrates Polygenic Risk Scores (PRS) as a foundational data modality, moving beyond traditional EHR-only approaches to build more holistic health profiles. Leveraging the extensive and diverse data from the All of Us (AoU) Research Program, this multimodal framework aims to learn complex relationships between clinical data and genetic predispositions. The methodology extends advancements in generative AI to the EHR foundation model space, enhancing predictive capabilities and interpretability. Evaluation on AoU data demonstrates the model's predictive value for the onset of various conditions, particularly Type 2 Diabetes (T2D), and illustrates the interplay between PRS and EHR data. The work also explores transfer learning for custom classification tasks, showcasing the architecture's versatility and efficiency. This approach is pivotal for unlocking new insights into disease prediction, proactive health management, risk stratification, and personalized treatment strategies, laying the groundwork for more personalized, equitable, and actionable real-world evidence generation in healthcare.

08 Dec 2025




Modern science advances fastest when thought meets action. LabOS represents the first AI co-scientist that unites computational reasoning with physical experimentation through multimodal perception, self-evolving agents, and Extended-Reality(XR)-enabled human-AI collaboration. By connecting multi-model AI agents, smart glasses, and robots, LabOS allows AI to see what scientists see, understand experimental context, and assist in real-time execution. Across applications -- from cancer immunotherapy target discovery to stem-cell engineering and material science -- LabOS shows that AI can move beyond computational design to participation, turning the laboratory into an intelligent, collaborative environment where human and machine discovery evolve together.

17 Oct 2025

Reliable, informative, and individual uncertainty quantification (UQ) remains missing in current ML community. This hinders the effective application of AI/ML to risk-sensitive domains. Most methods either fail to provide coverage on new data, inflate intervals so broadly that they are not actionable, or assign uncertainties that do not track actual error, especially under a distribution shift. In high-stakes drug discovery, protein-ligand affinity (PLI) prediction is especially challenging as assay noise is heterogeneous, chemical space is imbalanced and large, and practical evaluations routinely involve distribution shift. In this work, we introduce a novel uncertainty quantification method, Trustworthy Expert Split-conformal with Scaled Estimation for Efficient Reliable Adaptive intervals (TESSERA), that provides per-sample uncertainty with reliable coverage guarantee, informative and adaptive prediction interval widths that track the absolute error. We evaluate on protein-ligand binding affinity prediction under both independent and identically distributed (i.i.d.) and scaffold-based out-of-distribution (OOD) splits, comparing against strong UQ baselines. TESSERA attains near-nominal coverage and the best coverage-width trade-off as measured by the Coverage-Width Criterion (CWC), while maintaining competitive adaptivity (lowest Area Under the Sparsification Error (AUSE)). Size-Stratified Coverage (SSC) further confirms that intervals are right-sized, indicating width increases when data are scarce or noisy, and remain tight when predictions are reliable. By unifying Mixture of Expert (MoE) diversity with conformal calibration, TESSERA delivers trustworthy, tight, and adaptive uncertainties that are well-suited to selective prediction and downstream decision-making in the drug-discovery pipeline and other applications.

14 Oct 2025

mRNA design and optimization are important in synthetic biology and therapeutic development, but remain understudied in machine learning. Systematic optimization of mRNAs is hindered by the scarce and imbalanced data as well as complex sequence-function relationships. We present RNAGenScape, a property-guided manifold Langevin dynamics framework that iteratively updates mRNA sequences within a learned latent manifold. RNAGenScape combines an organized autoencoder, which structures the latent space by target properties for efficient and biologically plausible exploration, with a manifold projector that contracts each step of update back to the manifold. RNAGenScape supports property-guided optimization and smooth interpolation between sequences, while remaining robust under scarce and undersampled data, and ensuring that intermediate products are close to the viable mRNA manifold. Across three real mRNA datasets, RNAGenScape improves the target properties with high success rates and efficiency, outperforming various generative or optimization methods developed for proteins or non-biological data. By providing continuous, data-aligned trajectories that reveal how edits influence function, RNAGenScape establishes a scalable paradigm for controllable mRNA design and latent space exploration in mRNA sequence modeling.

13 Oct 2025
Researchers at Wuhan University, Hong Kong Polytechnic University, and Stanford University developed the "Protein-as-Second-Language" framework, enabling general large language models (LLMs) to interpret and reason about protein sequences in a zero-shot setting. This approach, which involves dynamically generating bilingual context from a curated 79,926 QA dataset, achieved an average 7% ROUGE-L improvement and consistently outperformed fine-tuned protein language models on various protein understanding tasks.

08 Oct 2025


SCPPDM, a denoising diffusion model, predicts single-cell gene expression responses to drug perturbations, including dose and chemical structure, demonstrating superior generalization to unseen drug-cell line combinations and novel drugs compared to prior methods. Its interpretable dual-knob guidance allows controlled prediction of drug effects while preserving cell-line identity.
There are no more papers matching your filters at the moment.
