
10 Dec 2025
Batch effects pose a significant challenge in the analysis of single-cell omics data, introducing technical artifacts that confound biological signals. While various computational methods have achieved empirical success in correcting these effects, they lack the formal theoretical guarantees required to assess their reliability and generalization. To bridge this gap, we introduce Mixture-Model-based Data Harmonization (MoDaH), a principled batch correction algorithm grounded in a rigorous statistical framework.
Under a new Gaussian-mixture-model with explicit parametrization of batch effects, we establish the minimax optimal error rates for batch correction and prove that MoDaH achieves this rate by leveraging the recent theoretical advances in clustering data from anisotropic Gaussian mixtures. This constitutes, to the best of our knowledge, the first theoretical guarantee for batch correction. Extensive experiments on diverse single-cell RNA-seq and spatial proteomics datasets demonstrate that MoDaH not only attains theoretical optimality but also achieves empirical performance comparable to or even surpassing those of state-of-the-art heuristics (e.g., Harmony, Seurat-V5, and LIGER), effectively balancing the removal of technical noise with the conservation of biological signal.

02 Dec 2025
Wastewater-based genomic surveillance has emerged as a powerful tool for population-level viral monitoring, offering comprehensive insights into circulating viral variants across entire communities. However, this approach faces significant computational challenges stemming from high sequencing noise, low viral coverage, fragmented reads, and the complete absence of labeled variant annotations. Traditional reference-based variant calling pipelines struggle with novel mutations and require extensive computational resources. We present a comprehensive framework for unsupervised viral variant detection using Vector-Quantized Variational Autoencoders (VQ-VAE) that learns discrete codebooks of genomic patterns from k-mer tokenized sequences without requiring reference genomes or variant labels. Our approach extends the base VQ-VAE architecture with masked reconstruction pretraining for robustness to missing data and contrastive learning for highly discriminative embeddings. Evaluated on SARS-CoV-2 wastewater sequencing data comprising approximately 100,000 reads, our VQ-VAE achieves 99.52% mean token-level accuracy and 56.33% exact sequence match rate while maintaining 19.73% codebook utilization (101 of 512 codes active), demonstrating efficient discrete representation learning. Contrastive fine-tuning with different projection dimensions yields substantial clustering improvements: 64-dimensional embeddings achieve +35% Silhouette score improvement (0.31 to 0.42), while 128-dimensional embeddings achieve +42% improvement (0.31 to 0.44), clearly demonstrating the impact of embedding dimensionality on variant discrimination capability. Our reference-free framework provides a scalable, interpretable approach to genomic surveillance with direct applications to public health monitoring.

28 Nov 2025
Machine- and deep-learning approaches for biological sequences depend critically on transforming raw DNA, RNA, and protein FASTA files into informative numerical representations. However, this process is often fragmented across multiple libraries and preprocessing steps, which creates a barrier for researchers without extensive computational expertise. To address this gap, we developed deepFEPS, an open-source toolkit that unifies state-of-the-art feature extraction methods for sequence data within a single, reproducible workflow. deepFEPS integrates five families of modern feature extractors - k-mer embeddings (Word2Vec, FastText), document-level embeddings (Doc2Vec), transformer-based encoders (DNABERT, ProtBERT, and ESM2), autoencoder-derived latent features, and graph-based embeddings - into one consistent platform. The system accepts FASTA input via a web interface or command-line tool, exposes key model parameters, and outputs analysis-ready feature matrices (CSV). Each run is accompanied by an automatic quality-control report including sequence counts, dimensionality, sparsity, variance distributions, class balance, and diagnostic visualizations. By consolidating advanced sequence embeddings into one environment, deepFEPS reduces preprocessing overhead, improves reproducibility, and shortens the path from raw sequences to downstream machine- and deep-learning applications. deepFEPS lowers the practical barrier to modern representation learning for bioinformatics, enabling both novice and expert users to generate advanced embeddings for classification, clustering, and predictive modeling. Its unified framework supports exploratory analyses, high-throughput studies, and integration into institutional workflows, while remaining extensible to emerging models and methods. The webserver is accessible at this https URL.
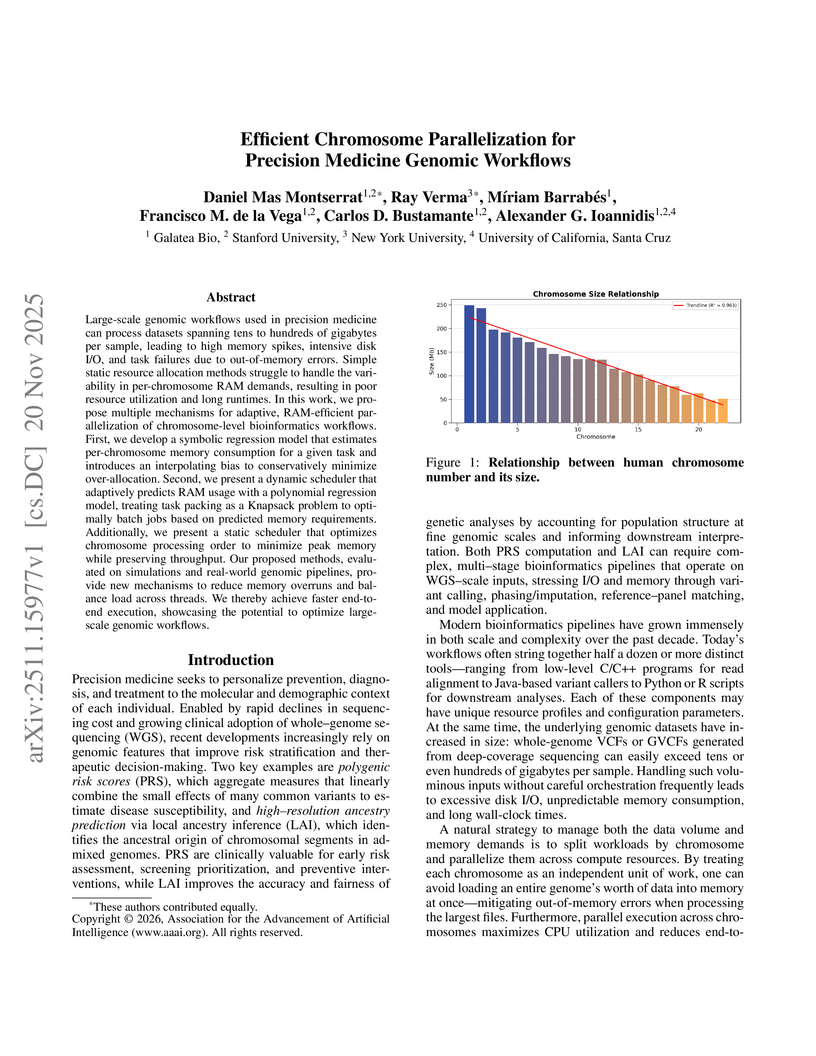
20 Nov 2025
Large-scale genomic workflows used in precision medicine can process datasets spanning tens to hundreds of gigabytes per sample, leading to high memory spikes, intensive disk I/O, and task failures due to out-of-memory errors. Simple static resource allocation methods struggle to handle the variability in per-chromosome RAM demands, resulting in poor resource utilization and long runtimes. In this work, we propose multiple mechanisms for adaptive, RAM-efficient parallelization of chromosome-level bioinformatics workflows. First, we develop a symbolic regression model that estimates per-chromosome memory consumption for a given task and introduces an interpolating bias to conservatively minimize over-allocation. Second, we present a dynamic scheduler that adaptively predicts RAM usage with a polynomial regression model, treating task packing as a Knapsack problem to optimally batch jobs based on predicted memory requirements. Additionally, we present a static scheduler that optimizes chromosome processing order to minimize peak memory while preserving throughput. Our proposed methods, evaluated on simulations and real-world genomic pipelines, provide new mechanisms to reduce memory overruns and balance load across threads. We thereby achieve faster end-to-end execution, showcasing the potential to optimize large-scale genomic workflows.

19 Nov 2025
Researchers from Wuhan University and Tsinghua University developed TDAM-CRC, a deep learning model that significantly improves prognostic stratification for colorectal cancer patients using whole-slide images, achieving a C-index of 0.747 on the TCGA cohort and outperforming existing methods. The study also uncovered molecular drivers, an immunosuppressive tumor microenvironment in high-risk tumors, and identified MRPL37 as an independent favorable prognostic biomarker, integrated into a robust clinical nomogram.

12 Nov 2025
Detecting chemical modifications on RNA molecules remains a key challenge in epitranscriptomics. Traditional reverse transcription-based sequencing methods introduce enzyme- and sequence-dependent biases and fragment RNA molecules, confounding the accurate mapping of modifications across the transcriptome. Nanopore direct RNA sequencing offers a powerful alternative by preserving native RNA molecules, enabling the detection of modifications at single-molecule resolution. However, current computational tools can identify only a limited subset of modification types within well-characterized sequence contexts for which ample training data exists. Here, we introduce a model-free computational method that reframes modification detection as an anomaly detection problem, requiring only canonical (unmodified) RNA reads without any other annotated data. For each nanopore read, our approach extracts robust, modification-sensitive features from the raw ionic current signal at a site using the signature transform, then computes an anomaly score by comparing the resulting feature vector to its nearest neighbors in an unmodified reference dataset. We convert anomaly scores into statistical p-values to enable anomaly detection at both individual read and site levels. Validation on densely-modified \textit{E. coli} rRNA demonstrates that our approach detects known sites harboring diverse modification types, without prior training on these modifications. We further applyied this framework to dengue virus (DENV) transcripts and mammalian mRNAs. For DENV sfRNA, it led to revealing a novel 2'-O-methylated site, which we validate orthogonally by qRT-PCR assays. These results demonstrate that our model-free approach operates robustly across different types of RNAs and datasets generated with different nanopore sequencing chemistries.

30 Sep 2025


Background: Large foundation models have revolutionized single-cell analysis, yet no kidney-specific model currently exists, and it remains unclear whether organ-focused models can outperform generalized models. The kidney's complex cellular architecture further complicate integration of large-scale omics data, where current frameworks trained on limited datasets struggle to correct batch effects, capture cross-modality variation, and generalize across species. Methods: We developed Nephrobase Cell+, the first kidney-focused large foundation model, pretrained on ~100 billion tokens from ~39.5 million single-cell and single-nucleus profiles across 4,319 samples. Nephrobase Cell+ uses a transformer-based encoder-decoder architecture with gene-token cross-attention and a mixture-of-experts module for scalable representation learning. Results: Nephrobase Cell+ sets a new benchmark for kidney single-cell analysis. It produces tightly clustered, biologically coherent embeddings in human and mouse kidneys, far surpassing previous foundation models such as Geneformer, scGPT, and UCE, as well as traditional methods such as PCA and autoencoders. It achieves the highest cluster concordance and batch-mixing scores, effectively removing donor/assay batch effects while preserving cell-type structure. Cross-species evaluation shows superior alignment of homologous cell types and >90% zero-shot annotation accuracy for major kidney lineages in both human and mouse. Even its 1B-parameter and 500M variants consistently outperform all existing models. Conclusions: Nephrobase Cell+ delivers a unified, high-fidelity representation of kidney biology that is robust, cross-species transferable, and unmatched by current single-cell foundation models, offering a powerful resource for kidney genomics and disease research.

23 Sep 2025
A framework called CEP-IP quantifies the explanatory power of individual cells in single-cell transcriptomics using optimized Generalized Additive Models, allowing identification of distinct prostate cancer cell subpopulations with varying TRPM4 and ribosomal gene expression patterns. The framework provides interpretable insights into biological processes related to cancer aggression and immune response.

15 Aug 2025
A fundamental limitation of probabilistic deep learning is its predominant reliance on Gaussian priors. This simplistic assumption prevents models from accurately capturing the complex, non-Gaussian landscapes of natural data, particularly in demanding domains like complex biological data, severely hindering the fidelity of the model for scientific discovery. The physically-grounded Boltzmann distribution offers a more expressive alternative, but it is computationally intractable on classical computers. To date, quantum approaches have been hampered by the insufficient qubit scale and operational stability required for the iterative demands of deep learning. Here, we bridge this gap by introducing the Quantum Boltzmann Machine-Variational Autoencoder (QBM-VAE), a large-scale and long-time stable hybrid quantum-classical architecture. Our framework leverages a quantum processor for efficient sampling from the Boltzmann distribution, enabling its use as a powerful prior within a deep generative model. Applied to million-scale single-cell datasets from multiple sources, the QBM-VAE generates a latent space that better preserves complex biological structures, consistently outperforming conventional Gaussian-based deep learning models like VAE and SCVI in essential tasks such as omics data integration, cell-type classification, and trajectory inference. It also provides a typical example of introducing a physics priori into deep learning to drive the model to acquire scientific discovery capabilities that breaks through data limitations. This work provides the demonstration of a practical quantum advantage in deep learning on a large-scale scientific problem and offers a transferable blueprint for developing hybrid quantum AI models.

12 Jul 2025
Transformers have revolutionized nucleotide sequence analysis, yet capturing long-range dependencies remains challenging. Recent studies show that autoregressive transformers often exhibit Markovian behavior by relying on fixed-length context windows for next-token prediction. However, standard self-attention mechanisms are computationally inefficient for long sequences due to their quadratic complexity and do not explicitly enforce global transition consistency.
We introduce CARMANIA (Context-Aware Regularization with Markovian Integration for Attention-Based Nucleotide Analysis), a self-supervised pretraining framework that augments next-token (NT) prediction with a transition-matrix (TM) loss. The TM loss aligns predicted token transitions with empirically derived n-gram statistics from each input sequence, encouraging the model to capture higher-order dependencies beyond local context. This integration enables CARMANIA to learn organism-specific sequence structures that reflect both evolutionary constraints and functional organization.
We evaluate CARMANIA across diverse genomic tasks, including regulatory element prediction, functional gene classification, taxonomic inference, antimicrobial resistance detection, and biosynthetic gene cluster classification. CARMANIA outperforms the previous best long-context model by at least 7 percent, matches state-of-the-art on shorter sequences (exceeding prior results on 20 out of 40 tasks while running approximately 2.5 times faster), and shows particularly strong improvements on enhancer and housekeeping gene classification tasks, including up to a 34 percent absolute gain in Matthews correlation coefficient (MCC) for enhancer prediction. The TM loss boosts accuracy in 33 of 40 tasks, especially where local motifs or regulatory patterns drive prediction.

25 May 2025


STFlow is a computational framework that predicts spatially-resolved gene expression directly from standard histology whole-slide images, eliminating the need for expensive wet-lab experiments. It leverages a flow matching approach to model joint gene expression distributions and achieved an 18% average relative improvement in prediction performance over pathology foundation models while demonstrating significantly enhanced computational efficiency.

26 Oct 2025


The advent of single-cell Assay for Transposase-Accessible Chromatin using sequencing (scATAC-seq) offers an innovative perspective for deciphering regulatory mechanisms by assembling a vast repository of single-cell chromatin accessibility data. While foundation models have achieved significant success in single-cell transcriptomics, there is currently no foundation model for scATAC-seq that supports zero-shot high-quality cell identification and comprehensive multi-omics analysis simultaneously. Key challenges lie in the high dimensionality and sparsity of scATAC-seq data, as well as the lack of a standardized schema for representing open chromatin regions (OCRs). Here, we present ChromFound, a foundation model tailored for scATAC-seq. ChromFound utilizes a hybrid architecture and genome-aware tokenization to effectively capture genome-wide long contexts and regulatory signals from dynamic chromatin landscapes. Pretrained on 1.97 million cells from 30 tissues and 6 disease conditions, ChromFound demonstrates broad applicability across 6 diverse tasks. Notably, it achieves robust zero-shot performance in generating universal cell representations and exhibits excellent transferability in cell type annotation and cross-omics prediction. By uncovering enhancer-gene links undetected by existing computational methods, ChromFound offers a promising framework for understanding disease risk variants in the noncoding genome.

20 Mar 2025


A novel neural network architecture called Lyra combines gated convolutions with state space models to achieve efficient biological sequence modeling, reducing parameter count by up to 120,000x compared to existing models while matching or exceeding their performance across protein, RNA, and CRISPR prediction tasks using just two GPUs.

01 Apr 2025




Advancements in DNA sequencing technologies have significantly improved our
ability to decode genomic sequences. However, the prediction and interpretation
of these sequences remain challenging due to the intricate nature of genetic
material. Large language models (LLMs) have introduced new opportunities for
biological sequence analysis. Recent developments in genomic language models
have underscored the potential of LLMs in deciphering DNA sequences.
Nonetheless, existing models often face limitations in robustness and
application scope, primarily due to constraints in model structure and training
data scale. To address these limitations, we present GENERator, a generative
genomic foundation model featuring a context length of 98k base pairs (bp) and
1.2B parameters. Trained on an expansive dataset comprising 386B bp of
eukaryotic DNA, the GENERator demonstrates state-of-the-art performance across
both established and newly proposed benchmarks. The model adheres to the
central dogma of molecular biology, accurately generating protein-coding
sequences that translate into proteins structurally analogous to known
families. It also shows significant promise in sequence optimization,
particularly through the prompt-responsive generation of enhancer sequences
with specific activity profiles. These capabilities position the GENERator as a
pivotal tool for genomic research and biotechnological advancement, enhancing
our ability to interpret and predict complex biological systems and enabling
precise genomic interventions. Implementation details and supplementary
resources are available at this https URL

04 Nov 2025

Research organisms provide invaluable insights into human biology and diseases, serving as essential tools for functional experiments, disease modeling, and drug testing. However, evolutionary divergence between humans and research organisms hinders effective knowledge transfer across species. Here, we review state-of-the-art methods for computationally transferring knowledge across species, primarily focusing on methods that utilize transcriptome data and/or molecular networks. Our review addresses four key areas: (1) transferring disease and gene annotation knowledge across species, (2) identifying functionally equivalent molecular components, (3) inferring equivalent perturbed genes or gene sets, and (4) identifying equivalent cell types. We conclude with an outlook on future directions and several key challenges that remain in cross-species knowledge transfer, including introducing the concept of "agnology" to describe functional equivalence of biological entities, regardless of their evolutionary origins. This concept is becoming pervasive in integrative data-driven models where evolutionary origins of functions can remain unresolved.

11 Aug 2025


We introduce RNA-FrameFlow, the first generative model for 3D RNA backbone design. We build upon SE(3) flow matching for protein backbone generation and establish protocols for data preparation and evaluation to address unique challenges posed by RNA modeling. We formulate RNA structures as a set of rigid-body frames and associated loss functions which account for larger, more conformationally flexible RNA backbones (13 atoms per nucleotide) vs. proteins (4 atoms per residue). Toward tackling the lack of diversity in 3D RNA datasets, we explore training with structural clustering and cropping augmentations. Additionally, we define a suite of evaluation metrics to measure whether the generated RNA structures are globally self-consistent (via inverse folding followed by forward folding) and locally recover RNA-specific structural descriptors. The most performant version of RNA-FrameFlow generates locally realistic RNA backbones of 40-150 nucleotides, over 40% of which pass our validity criteria as measured by a self-consistency TM-score >= 0.45, at which two RNAs have the same global fold. Open-source code: this https URL

18 Mar 2024


Researchers from Northwestern University and Stony Brook University developed DNABERT-2, an efficient genome foundation model, by introducing Byte Pair Encoding and Attention with Linear Biases for improved performance and computational accessibility. The model achieves competitive results on a new multi-species benchmark across various genomic tasks while significantly reducing resource requirements compared to prior state-of-the-art models.

14 Nov 2023


HyenaDNA is a genomic foundation model that processes DNA sequences up to 1 million tokens at single nucleotide resolution, addressing the long-range context limitations of prior models. The model achieves state-of-the-art performance across various genomic benchmarks and demonstrates the first application of in-context learning in genomics, while being significantly faster than Transformer models.

12 Oct 2020

Researchers developed deep learning models to predict adverse drug reactions by integrating rat liver toxicogenomics data with human adverse event reports, achieving a mean prediction accuracy of 85.71% across various ADR types. The approach also identified specific gene signatures associated with predicted ADRs, providing biological interpretability.

22 Feb 2024

At the last step of short read mapping, the candidate locations of the reads
on the reference genome are verified to compute their differences from the
corresponding reference segments using sequence alignment algorithms.
Calculating the similarities and differences between two sequences is still
computationally expensive since approximate string matching techniques
traditionally inherit dynamic programming algorithms with quadratic time and
space complexity. We introduce GateKeeper-GPU, a fast and accurate
pre-alignment filter that efficiently reduces the need for expensive sequence
alignment. GateKeeper-GPU provides two main contributions: first, improving the
filtering accuracy of GateKeeper (a lightweight pre-alignment filter), and
second, exploiting the massive parallelism provided by the large number of GPU
threads of modern GPUs to examine numerous sequence pairs rapidly and
concurrently. By reducing the work, GateKeeper-GPU provides an acceleration of
2.9x to sequence alignment and up to 1.4x speedup to the end-to-end execution
time of a comprehensive read mapper (mrFAST). GateKeeper-GPU is available at
https://github.com/BilkentCompGen/GateKeeper-GPU.
There are no more papers matching your filters at the moment.
