
13 Sep 2024
We explore the application of Vision Transformer (ViT) for handwritten text recognition. The limited availability of labeled data in this domain poses challenges for achieving high performance solely relying on ViT. Previous transformer-based models required external data or extensive pre-training on large datasets to excel. To address this limitation, we introduce a data-efficient ViT method that uses only the encoder of the standard transformer. We find that incorporating a Convolutional Neural Network (CNN) for feature extraction instead of the original patch embedding and employ Sharpness-Aware Minimization (SAM) optimizer to ensure that the model can converge towards flatter minima and yield notable enhancements. Furthermore, our introduction of the span mask technique, which masks interconnected features in the feature map, acts as an effective regularizer. Empirically, our approach competes favorably with traditional CNN-based models on small datasets like IAM and READ2016. Additionally, it establishes a new benchmark on the LAM dataset, currently the largest dataset with 19,830 training text lines. The code is publicly available at: this https URL.

26 Sep 2025
We consider the problem of computing persistent homology (PH) for large-scale Euclidean point cloud data, aimed at downstream machine learning tasks, where the exponential growth of the most widely-used Vietoris-Rips complex imposes serious computational limitations. Although more scalable alternatives such as the Alpha complex or sparse Rips approximations exist, they often still result in a prohibitively large number of simplices. This poses challenges in the complex construction and in the subsequent PH computation, prohibiting their use on large-scale point clouds. To mitigate these issues, we introduce the Flood complex, inspired by the advantages of the Alpha and Witness complex constructions. Informally, at a given filtration value , the Flood complex contains all simplices from a Delaunay triangulation of a small subset of the point cloud that are fully covered by balls of radius emanating from , a process we call flooding. Our construction allows for efficient PH computation, possesses several desirable theoretical properties, and is amenable to GPU parallelization. Scaling experiments on 3D point cloud data show that we can compute PH of up to dimension 2 on several millions of points. Importantly, when evaluating object classification performance on real-world and synthetic data, we provide evidence that this scaling capability is needed, especially if objects are geometrically or topologically complex, yielding performance superior to other PH-based methods and neural networks for point cloud data.

19 Sep 2025
AUTOGRAPH, an autoregressive model from the Max Planck Institute of Biochemistry, transforms graph generation into a sequence prediction task for decoder-only transformers. The method achieves up to 100x faster generation and 3x faster training than leading diffusion models, demonstrating state-of-the-art performance on molecular graphs and robust scalability.

07 Oct 2025
Existing methods for evaluating graph generative models primarily rely on Maximum Mean Discrepancy (MMD) metrics based on graph descriptors. While these metrics can rank generative models, they do not provide an absolute measure of performance. Their values are also highly sensitive to extrinsic parameters, namely kernel and descriptor parametrization, making them incomparable across different graph descriptors. We introduce PolyGraph Discrepancy (PGD), a new evaluation framework that addresses these limitations. It approximates the Jensen-Shannon distance of graph distributions by fitting binary classifiers to distinguish between real and generated graphs, featurized by these descriptors. The data log-likelihood of these classifiers approximates a variational lower bound on the JS distance between the two distributions. Resulting metrics are constrained to the unit interval [0,1] and are comparable across different graph descriptors. We further derive a theoretically grounded summary metric that combines these individual metrics to provide a maximally tight lower bound on the distance for the given descriptors. Thorough experiments demonstrate that PGD provides a more robust and insightful evaluation compared to MMD metrics. The PolyGraph framework for benchmarking graph generative models is made publicly available at this https URL.

07 Oct 2024
Message-passing graph neural networks (GNNs) excel at capturing local relationships but struggle with long-range dependencies in graphs. In contrast, graph transformers (GTs) enable global information exchange but often oversimplify the graph structure by representing graphs as sets of fixed-length vectors. This work introduces a novel architecture that overcomes the shortcomings of both approaches by combining the long-range information of random walks with local message passing. By treating random walks as sequences, our architecture leverages recent advances in sequence models to effectively capture long-range dependencies within these walks. Based on this concept, we propose a framework that offers (1) more expressive graph representations through random walk sequences, (2) the ability to utilize any sequence model for capturing long-range dependencies, and (3) the flexibility by integrating various GNN and GT architectures. Our experimental evaluations demonstrate that our approach achieves significant performance improvements on 19 graph and node benchmark datasets, notably outperforming existing methods by up to 13\% on the PascalVoc-SP and COCO-SP datasets. The code is available at this https URL.

22 Oct 2025
The relationship between RNA structure and function has recently attracted interest within the deep learning community, a trend expected to intensify as nucleic acid structure models advance. Despite this momentum, the lack of standardized, accessible benchmarks for applying deep learning to RNA 3D structures hinders progress. To this end, we introduce a collection of seven benchmarking datasets specifically designed to support RNA structure-function prediction. Built on top of the established Python package rnaglib, our library streamlines data distribution and encoding, provides tools for dataset splitting and evaluation, and offers a comprehensive, user-friendly environment for model comparison. The modular and reproducible design of our datasets encourages community contributions and enables rapid customization. To demonstrate the utility of our benchmarks, we report baseline results for all tasks using a relational graph neural network.

30 Sep 2025
We introduce InVirtuoGen, a discrete flow generative model for fragmented SMILES for de novo and fragment-constrained generation, and target-property/lead optimization of small molecules. The model learns to transform a uniform source over all possible tokens into the data distribution. Unlike masked models, its training loss accounts for predictions on all sequence positions at every denoising step, shifting the generation paradigm from completion to refinement, and decoupling the number of sampling steps from the sequence length. For \textit{de novo} generation, InVirtuoGen achieves a stronger quality-diversity pareto frontier than prior fragment-based models and competitive performance on fragment-constrained tasks. For property and lead optimization, we propose a hybrid scheme that combines a genetic algorithm with a Proximal Property Optimization fine-tuning strategy adapted to discrete flows. Our approach sets a new state-of-the-art on the Practical Molecular Optimization benchmark, measured by top-10 AUC across tasks, and yields higher docking scores in lead optimization than previous baselines. InVirtuoGen thus establishes a versatile generative foundation for drug discovery, from early hit finding to multi-objective lead optimization. We further contribute to open science by releasing pretrained checkpoints and code, making our results fully reproducible\footnote{this https URL}.

04 Feb 2025

Graph generative models often face a critical trade-off between learning complex distributions and achieving fast generation speed. We introduce Autoregressive Noisy Filtration Modeling (ANFM), a novel approach that addresses both challenges. ANFM leverages filtration, a concept from topological data analysis, to transform graphs into short sequences of monotonically increasing subgraphs. This formulation extends the sequence families used in previous autoregressive models. To learn from these sequences, we propose a novel autoregressive graph mixer model. Our experiments suggest that exposure bias might represent a substantial hurdle in autoregressive graph generation and we introduce two mitigation strategies to address it: noise augmentation and a reinforcement learning approach. Incorporating these techniques leads to substantial performance gains, making ANFM competitive with state-of-the-art diffusion models across diverse synthetic and real-world datasets. Notably, ANFM produces remarkably short sequences, achieving a 100-fold speedup in generation time compared to diffusion models. This work marks a significant step toward high-throughput graph generation.

24 Dec 2021
A complex balanced kinetic system is absolutely complex balanced (ACB) if every positive equilibrium is complex balanced. Two results on absolute complex balancing were foundational for modern chemical reaction network theory (CRNT): in 1972, M. Feinberg proved that any deficiency zero complex balanced system is absolutely complex balanced. In the same year, F. Horn and R. Jackson showed that the (full) converse of the result is not true: any complex balanced mass action system, regardless of its deficiency, is absolutely complex balanced. In this paper, we present initial results on the extension of the Horn and Jackson ACB Theorem. In particular, we focus on other kinetic systems with positive deficiency where complex balancing implies absolute complex balancing. While doing so, we found out that complex balanced power law reactant determined kinetic systems (PL-RDK) systems are not ACB. In our search for necessary and sufficient conditions for complex balanced systems to be absolutely complex balanced, we came across the so-called CLP systems (complex balanced systems with a desired "log parametrization" property). It is shown that complex balanced systems with bi-LP property are absolutely complex balanced. For non-CLP systems, we discuss novel methods for finding sufficient conditions for ACB in kinetic systems containing non-CLP systems: decompositions, the Positive Function Factor (PFF) and the Coset Intersection Count (CIC) and their application to poly-PL and Hill-type systems.
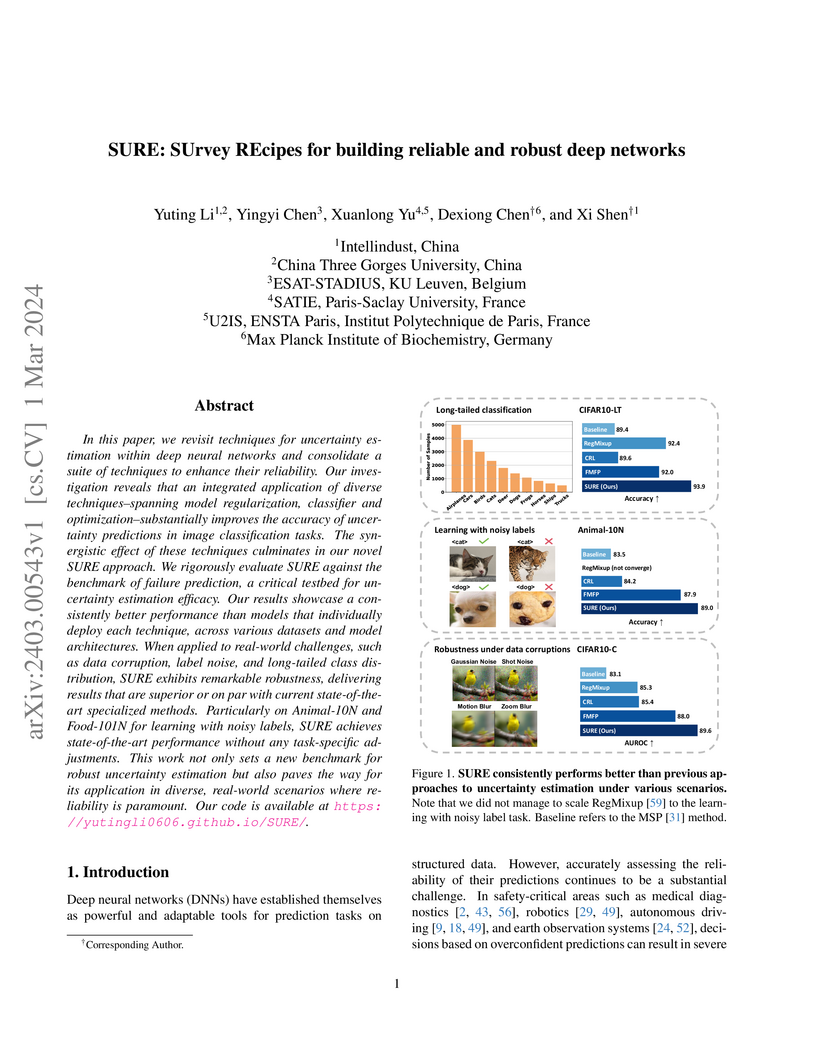
01 Mar 2024

In this paper, we revisit techniques for uncertainty estimation within deep neural networks and consolidate a suite of techniques to enhance their reliability. Our investigation reveals that an integrated application of diverse techniques--spanning model regularization, classifier and optimization--substantially improves the accuracy of uncertainty predictions in image classification tasks. The synergistic effect of these techniques culminates in our novel SURE approach. We rigorously evaluate SURE against the benchmark of failure prediction, a critical testbed for uncertainty estimation efficacy. Our results showcase a consistently better performance than models that individually deploy each technique, across various datasets and model architectures. When applied to real-world challenges, such as data corruption, label noise, and long-tailed class distribution, SURE exhibits remarkable robustness, delivering results that are superior or on par with current state-of-the-art specialized methods. Particularly on Animal-10N and Food-101N for learning with noisy labels, SURE achieves state-of-the-art performance without any task-specific adjustments. This work not only sets a new benchmark for robust uncertainty estimation but also paves the way for its application in diverse, real-world scenarios where reliability is paramount. Our code is available at \url{this https URL}.

29 Apr 2025
In this work, we theoretically demonstrate that current graph positional
encodings (PEs) are not beneficial and could potentially hurt performance in
tasks involving heterophilous graphs, where nodes that are close tend to have
different labels. This limitation is critical as many real-world networks
exhibit heterophily, and even highly homophilous graphs can contain local
regions of strong heterophily. To address this limitation, we propose Learnable
Laplacian Positional Encodings (LLPE), a new PE that leverages the full
spectrum of the graph Laplacian, enabling them to capture graph structure on
both homophilous and heterophilous graphs. Theoretically, we prove LLPE's
ability to approximate a general class of graph distances and demonstrate its
generalization properties. Empirically, our evaluation on 12 benchmarks
demonstrates that LLPE improves accuracy across a variety of GNNs, including
graph transformers, by up to 35% and 14% on synthetic and real-world graphs,
respectively. Going forward, our work represents a significant step towards
developing PEs that effectively capture complex structures in heterophilous
graphs.

21 Feb 2025
 University of Waterloo
University of Waterloo Monash University
Monash University Chinese Academy of Sciences
Chinese Academy of Sciences Sun Yat-Sen University
Sun Yat-Sen University Fudan UniversityIndiana University
Fudan UniversityIndiana University Westlake University
Westlake University ETH Zürich
ETH Zürich University of California, San Diego
University of California, San Diego Peking University
Peking University University of British ColumbiaHarvard Medical SchoolPengcheng Laboratory
University of British ColumbiaHarvard Medical SchoolPengcheng Laboratory University of Sydney
University of Sydney TencentMax Planck Institute of BiochemistryTulane UniversityDana-Farber Cancer InstituteBrigham Young UniversityThermo Fisher ScientificUniversity of AntwerpBristol Myers SquibbCharité – Universitätsmedizin BerlinAI for Science InstituteEuropean Molecular Biology LaboratoryBeijing Institute of LifeomicsInstitute for Systems BiologyBruker
TencentMax Planck Institute of BiochemistryTulane UniversityDana-Farber Cancer InstituteBrigham Young UniversityThermo Fisher ScientificUniversity of AntwerpBristol Myers SquibbCharité – Universitätsmedizin BerlinAI for Science InstituteEuropean Molecular Biology LaboratoryBeijing Institute of LifeomicsInstitute for Systems BiologyBrukerArtificial intelligence (AI) is transforming scientific research, including
proteomics. Advances in mass spectrometry (MS)-based proteomics data quality,
diversity, and scale, combined with groundbreaking AI techniques, are unlocking
new challenges and opportunities in biological discovery. Here, we highlight
key areas where AI is driving innovation, from data analysis to new biological
insights. These include developing an AI-friendly ecosystem for proteomics data
generation, sharing, and analysis; improving peptide and protein identification
and quantification; characterizing protein-protein interactions and protein
complexes; advancing spatial and perturbation proteomics; integrating
multi-omics data; and ultimately enabling AI-empowered virtual cells.

26 Sep 2025
We consider the problem of computing persistent homology (PH) for large-scale Euclidean point cloud data, aimed at downstream machine learning tasks, where the exponential growth of the most widely-used Vietoris-Rips complex imposes serious computational limitations. Although more scalable alternatives such as the Alpha complex or sparse Rips approximations exist, they often still result in a prohibitively large number of simplices. This poses challenges in the complex construction and in the subsequent PH computation, prohibiting their use on large-scale point clouds. To mitigate these issues, we introduce the Flood complex, inspired by the advantages of the Alpha and Witness complex constructions. Informally, at a given filtration value , the Flood complex contains all simplices from a Delaunay triangulation of a small subset of the point cloud that are fully covered by balls of radius emanating from , a process we call flooding. Our construction allows for efficient PH computation, possesses several desirable theoretical properties, and is amenable to GPU parallelization. Scaling experiments on 3D point cloud data show that we can compute PH of up to dimension 2 on several millions of points. Importantly, when evaluating object classification performance on real-world and synthetic data, we provide evidence that this scaling capability is needed, especially if objects are geometrically or topologically complex, yielding performance superior to other PH-based methods and neural networks for point cloud data.

10 Apr 2015

As a follow-up to the highly-cited authors list published by Thomson Reuters
in June 2014, we analyze the top-1% most frequently cited papers published
between 2002 and 2012 included in the Web of Science (WoS) subject category
"Information Science & Library Science." 798 authors contributed to 305 top-1%
publications; these authors were employed at 275 institutions. The authors at
Harvard University contributed the largest number of papers, when the addresses
are whole-number counted. However, Leiden University leads the ranking, if
fractional counting is used.
Twenty-three of the 798 authors were also listed as most highly-cited authors
by Thomson Reuters in June 2014 (this http URL). Twelve of these 23
authors were involved in publishing four or more of the 305 papers under study.
Analysis of co-authorship relations among the 798 highly-cited scientists shows
that co-authorships are based on common interests in a specific topic. Three
topics were important between 2002 and 2012: (1) collection and exploitation of
information in clinical practices, (2) the use of internet in public
communication and commerce, and (3) scientometrics.

21 Jul 2024
LMU MunichFriedrich-Alexander-Universität Erlangen-Nürnberg Technical University of MunichMax Planck Institute of BiochemistryUniversity of HamburgUniversity of Southern DenmarkHelmholtz Center MunichLeiden University Medical CenterUniversity Hospital ErlangenUniversity of GreifswaldGerman Center for Neurodegenerative DiseasesFraunhofer Institute for Cell Therapy and Immunology IZIUniversity of Applied Science CoburgGerman Center for Cardiovascular Diseases
Technical University of MunichMax Planck Institute of BiochemistryUniversity of HamburgUniversity of Southern DenmarkHelmholtz Center MunichLeiden University Medical CenterUniversity Hospital ErlangenUniversity of GreifswaldGerman Center for Neurodegenerative DiseasesFraunhofer Institute for Cell Therapy and Immunology IZIUniversity of Applied Science CoburgGerman Center for Cardiovascular Diseases
Technical University of MunichMax Planck Institute of BiochemistryUniversity of HamburgUniversity of Southern DenmarkHelmholtz Center MunichLeiden University Medical CenterUniversity Hospital ErlangenUniversity of GreifswaldGerman Center for Neurodegenerative DiseasesFraunhofer Institute for Cell Therapy and Immunology IZIUniversity of Applied Science CoburgGerman Center for Cardiovascular DiseasesQuantitative mass spectrometry has revolutionized proteomics by enabling
simultaneous quantification of thousands of proteins. Pooling patient-derived
data from multiple institutions enhances statistical power but raises
significant privacy concerns. Here we introduce FedProt, the first
privacy-preserving tool for collaborative differential protein abundance
analysis of distributed data, which utilizes federated learning and additive
secret sharing. In the absence of a multicenter patient-derived dataset for
evaluation, we created two, one at five centers from LFQ E.coli experiments and
one at three centers from TMT human serum. Evaluations using these datasets
confirm that FedProt achieves accuracy equivalent to DEqMS applied to pooled
data, with completely negligible absolute differences no greater than \text{4
\times 10^{-12}}. In contrast, -log10(p-values) computed by the most accurate
meta-analysis methods diverged from the centralized analysis results by up to
25-27. FedProt is available as a web tool with detailed documentation as a
FeatureCloud App.

17 May 2024
Understanding the insulin signaling cascade provides insights on the underlying mechanisms of biological phenomena such as insulin resistance, diabetes, Alzheimer's disease, and cancer. For this reason, previous studies utilized chemical reaction network theory to perform comparative analyses of reaction networks of insulin signaling in healthy (INSMS: INSulin Metabolic Signaling) and diabetic cells (INRES: INsulin RESistance). This study extends these analyses using various methods which give further insights regarding insulin signaling. Using embedded networks, we discuss evidence of the presence of a structural "bifurcation" in the signaling process between INSMS and INRES. Concordance profiles of INSMS and INRES show that both have a high propensity to remain monostationary. Moreover, the concordance properties allow us to present heuristic evidence that INRES has a higher level of stability beyond its monostationarity. Finally, we discuss a new way of analyzing reaction networks through network translation. This method gives rise to three new insights: (i) each stoichiometric class of INSMS and INRES contains a unique positive equilibrium; (ii) any positive equilibrium of INSMS is exponentially stable and is a global attractor in its stoichiometric class; and (iii) any positive equilibrium of INRES is locally asymptotically stable. These results open up opportunities for collaboration with experimental biologists to understand insulin signaling better.

21 Apr 2025

Comparing mathematical models offers a means to evaluate competing scientific
theories. However, exact methods of model calibration are not applicable to
many probabilistic models which simulate high-dimensional spatio-temporal data.
Approximate Bayesian Computation is a widely-used method for parameter
inference and model selection in such scenarios, and it may be combined with
Topological Data Analysis to study models which simulate data with fine spatial
structure. We develop a flexible pipeline for parameter inference and model
selection in spatio-temporal models. Our pipeline identifies topological
summary statistics which quantify spatio-temporal data and uses them to
approximate parameter and model posterior distributions. We validate our
pipeline on models of tumour-induced angiogenesis, inferring four parameters in
three established models and identifying the correct model in synthetic
test-cases.

20 Nov 2024
The multistationarity or the existence of steady-state multiplicity in the Earth System raises the possibility that the Earth may reach a "tipping point" and rapidly transition to a warmer steady-state from which recovery may be practically impossible. In detailed Earth models that require extensive computation time, it is difficult to make an a priori prediction of the possibility of multistationarity. In this study, we demonstrate Chemical Reaction Network Theory (CRNT) analysis of a simple heuristic box model of the Earth System carbon cycle with the human intervention of Direct Air Capture. The analysis reveals necessary conditions for the combination of system parameters where steady-state multiplicity may exist. With this method, other negative emissions technologies (NET) may be screened in a relatively simple manner to aid in the priority setting by policymakers.

30 May 2024
Maternal investment directly shapes early developmental conditions and therefore has longterm fitness consequences for the offspring. In oviparous species prenatal maternal investment is fixed at the time of laying. To ensure the best survival chances for most of their offspring, females must equip their eggs with the resources required to perform well under various circumstances, yet the actual mechanisms remain unknown. Here we describe the blue tit egg albumen and yolk proteomes and evaluate their potential to mediate maternal effects. We show that variation in egg composition (proteins, lipids, carotenoids) primarily depends on laying order and female age. Egg proteomic profiles are mainly driven by laying order, and investment in the egg proteome is functionally biased among eggs. Our results suggest that maternal effects on egg composition result from both passive and active (partly compensatory) mechanisms, and that variation in egg composition creates diverse biochemical environments for embryonic development.

03 May 2024

Understanding the connection between complex structural features of RNA and
biological function is a fundamental challenge in evolutionary studies and in
RNA design. However, building datasets of RNA 3D structures and making
appropriate modeling choices remains time-consuming and lacks standardization.
In this chapter, we describe the use of rnaglib, to train supervised and
unsupervised machine learning-based function prediction models on datasets of
RNA 3D structures.
There are no more papers matching your filters at the moment.
