
10 Aug 2024
Undesignable RNA Structure Identification via Rival Structure Generation and Structure Decomposition
Undesignable RNA Structure Identification via Rival Structure Generation and Structure Decomposition
RNA design is the search for a sequence or set of sequences that will fold into predefined structures, also known as the inverse problem of RNA folding. While numerous RNA design methods have been invented to find sequences capable of folding into a target structure, little attention has been given to the identification of undesignable structures according to the minimum free energy (MFE) criterion under the Turner model. In this paper, we address this gap by first introducing mathematical theorems outlining sufficient conditions for recognizing undesignable structures, then proposing efficient algorithms, guided by these theorems, to verify the undesignability of RNA structures. Through the application of these theorems and algorithms to the Eterna100 puzzles, we demonstrate the ability to efficiently establish that 15 of the puzzles indeed fall within the category of undesignable structures. In addition, we provide specific insights from the study of undesignability, in the hope that it will enable more understanding of RNA folding and RNA design.

28 May 2024
Heidelberg University UCLAUniversity of ZurichGeorge Washington University
UCLAUniversity of ZurichGeorge Washington University University of California, Irvine
University of California, Irvine Stanford University
Stanford University University of Michigan
University of Michigan Cornell University
Cornell University University of California, San Diego
University of California, San Diego McGill University
McGill University Northwestern UniversityUniversity of Missouri
Northwestern UniversityUniversity of Missouri University of PennsylvaniaMassachusetts General HospitalUppsala University
University of PennsylvaniaMassachusetts General HospitalUppsala University Duke UniversityHelmholtz MunichMayo ClinicWeill Cornell MedicineIran University of Medical SciencesChildren’s National HospitalUniversity Hospital UlmNational Institute of HealthSage BionetworksUniversity of Rochester Medical CenterMercy Catholic Medical CenterKlinikum rechts der Isar, Technical University of MunichThe Children’s Hospital of PhiladelphiaC.M.H. Lahore Medical College & Institute of DentistryRoss University School of MedicineNassau Medical CenterUniversity of LavalIndiana UniveristyHospital Garcia de Orta - Unidade Local de Sa ́ude de Almada Seixal, EPEScripps Clinic Medical GroupHospital da Luz LisboaVA San Diego Medical CenterASST Ovest Milanese - Legnano
Duke UniversityHelmholtz MunichMayo ClinicWeill Cornell MedicineIran University of Medical SciencesChildren’s National HospitalUniversity Hospital UlmNational Institute of HealthSage BionetworksUniversity of Rochester Medical CenterMercy Catholic Medical CenterKlinikum rechts der Isar, Technical University of MunichThe Children’s Hospital of PhiladelphiaC.M.H. Lahore Medical College & Institute of DentistryRoss University School of MedicineNassau Medical CenterUniversity of LavalIndiana UniveristyHospital Garcia de Orta - Unidade Local de Sa ́ude de Almada Seixal, EPEScripps Clinic Medical GroupHospital da Luz LisboaVA San Diego Medical CenterASST Ovest Milanese - Legnano
UCLAUniversity of ZurichGeorge Washington UniversityUniversity of California, IrvineStanford UniversityUniversity of MichiganCornell UniversityUniversity of California, San DiegoMcGill UniversityNorthwestern UniversityUniversity of MissouriUniversity of PennsylvaniaMassachusetts General HospitalUppsala UniversityDuke UniversityHelmholtz MunichMayo ClinicWeill Cornell MedicineIran University of Medical SciencesChildren’s National HospitalUniversity Hospital UlmNational Institute of HealthSage BionetworksUniversity of Rochester Medical CenterMercy Catholic Medical CenterKlinikum rechts der Isar, Technical University of MunichThe Children’s Hospital of PhiladelphiaC.M.H. Lahore Medical College & Institute of DentistryRoss University School of MedicineNassau Medical CenterUniversity of LavalIndiana UniveristyHospital Garcia de Orta - Unidade Local de Sa ́ude de Almada Seixal, EPEScripps Clinic Medical GroupHospital da Luz LisboaVA San Diego Medical CenterASST Ovest Milanese - LegnanoGliomas are the most common malignant primary brain tumors in adults and one of the deadliest types of cancer. There are many challenges in treatment and monitoring due to the genetic diversity and high intrinsic heterogeneity in appearance, shape, histology, and treatment response. Treatments include surgery, radiation, and systemic therapies, with magnetic resonance imaging (MRI) playing a key role in treatment planning and post-treatment longitudinal assessment. The 2024 Brain Tumor Segmentation (BraTS) challenge on post-treatment glioma MRI will provide a community standard and benchmark for state-of-the-art automated segmentation models based on the largest expert-annotated post-treatment glioma MRI dataset. Challenge competitors will develop automated segmentation models to predict four distinct tumor sub-regions consisting of enhancing tissue (ET), surrounding non-enhancing T2/fluid-attenuated inversion recovery (FLAIR) hyperintensity (SNFH), non-enhancing tumor core (NETC), and resection cavity (RC). Models will be evaluated on separate validation and test datasets using standardized performance metrics utilized across the BraTS 2024 cluster of challenges, including lesion-wise Dice Similarity Coefficient and Hausdorff Distance. Models developed during this challenge will advance the field of automated MRI segmentation and contribute to their integration into clinical practice, ultimately enhancing patient care.

03 Jun 2025
 University of Washington
University of Washington Harvard UniversityUniversity of UtahStanford UniversityMcGill UniversityHarvard Medical SchoolMassachusetts General HospitalUniversity of Tübingen
Harvard UniversityUniversity of UtahStanford UniversityMcGill UniversityHarvard Medical SchoolMassachusetts General HospitalUniversity of Tübingen The Ohio State UniversityDana-Farber Cancer InstituteAgency for Science Technology and Research (A*STAR)Brigham and Women’s HospitalHelmholtz Center MunichOregon Health & Science UniversityBroad Institute of Harvard and MITHarvard-MITUniversity of Rochester Medical CenterARUP Institute for Clinical and Experimental PathologyBeth-Israel Deaconess Medical Center
The Ohio State UniversityDana-Farber Cancer InstituteAgency for Science Technology and Research (A*STAR)Brigham and Women’s HospitalHelmholtz Center MunichOregon Health & Science UniversityBroad Institute of Harvard and MITHarvard-MITUniversity of Rochester Medical CenterARUP Institute for Clinical and Experimental PathologyBeth-Israel Deaconess Medical CenterKRONOS introduces the first foundation model specifically designed for spatial proteomics, leveraging a massive dataset of 47 million image patches to learn generalizable representations. This model enables superior cell phenotyping, facilitates robust segmentation-free analysis, and improves patient stratification and image retrieval across diverse experimental conditions and tissue types.
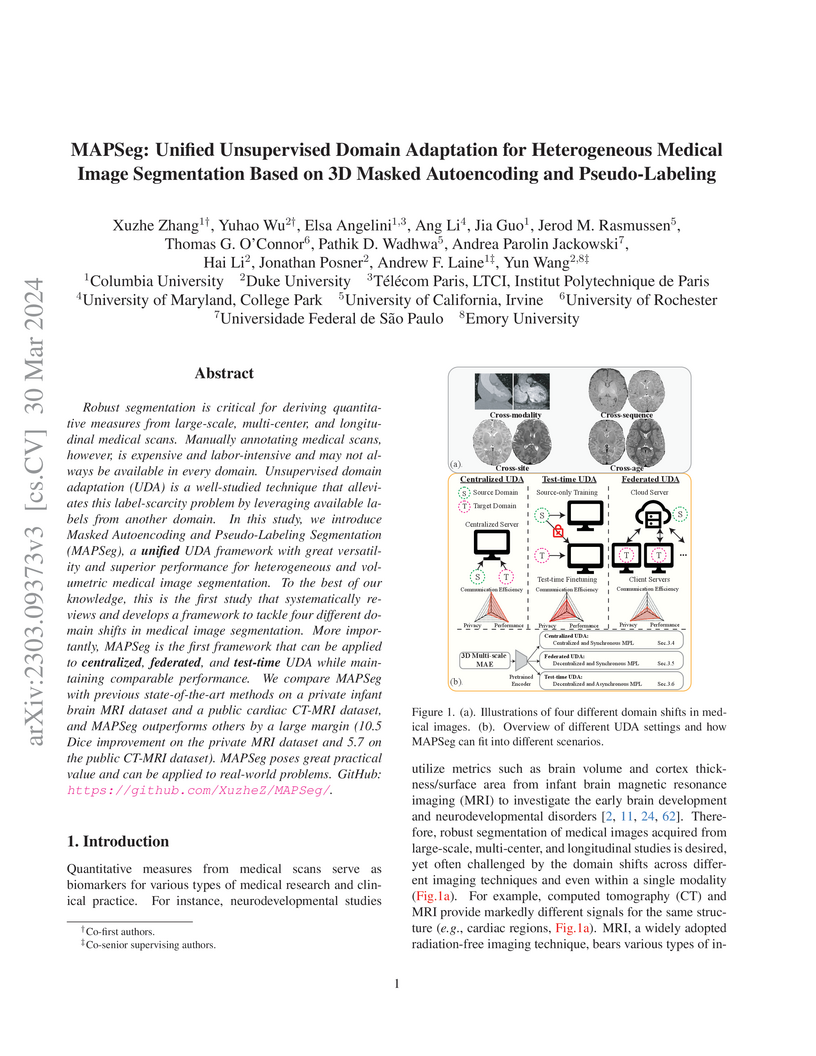
30 Mar 2024


Robust segmentation is critical for deriving quantitative measures from large-scale, multi-center, and longitudinal medical scans. Manually annotating medical scans, however, is expensive and labor-intensive and may not always be available in every domain. Unsupervised domain adaptation (UDA) is a well-studied technique that alleviates this label-scarcity problem by leveraging available labels from another domain. In this study, we introduce Masked Autoencoding and Pseudo-Labeling Segmentation (MAPSeg), a UDA framework with great versatility and superior performance for heterogeneous and volumetric medical image segmentation. To the best of our knowledge, this is the first study that systematically reviews and develops a framework to tackle four different domain shifts in medical image segmentation. More importantly, MAPSeg is the first framework that can be applied to , , and UDA while maintaining comparable performance. We compare MAPSeg with previous state-of-the-art methods on a private infant brain MRI dataset and a public cardiac CT-MRI dataset, and MAPSeg outperforms others by a large margin (10.5 Dice improvement on the private MRI dataset and 5.7 on the public CT-MRI dataset). MAPSeg poses great practical value and can be applied to real-world problems. GitHub: this https URL.

26 Apr 2025

Limited accessibility to neurological care leads to underdiagnosed
Parkinson's Disease (PD), preventing early intervention. Existing AI-based PD
detection methods primarily focus on unimodal analysis of motor or speech
tasks, overlooking the multifaceted nature of the disease. To address this, we
introduce a large-scale, multi-task video dataset consisting of 1102 sessions
(each containing videos of finger tapping, facial expression, and speech tasks
captured via webcam) from 845 participants (272 with PD). We propose a novel
Uncertainty-calibrated Fusion Network (UFNet) that leverages this multimodal
data to enhance diagnostic accuracy. UFNet employs independent task-specific
networks, trained with Monte Carlo Dropout for uncertainty quantification,
followed by self-attended fusion of features, with attention weights
dynamically adjusted based on task-specific uncertainties. To ensure
patient-centered evaluation, the participants were randomly split into three
sets: 60% for training, 20% for model selection, and 20% for final performance
evaluation. UFNet significantly outperformed single-task models in terms of
accuracy, area under the ROC curve (AUROC), and sensitivity while maintaining
non-inferior specificity. Withholding uncertain predictions further boosted the
performance, achieving 88.0+-0.3%$ accuracy, 93.0+-0.2% AUROC, 79.3+-0.9%
sensitivity, and 92.6+-0.3% specificity, at the expense of not being able to
predict for 2.3+-0.3% data (+- denotes 95% confidence interval). Further
analysis suggests that the trained model does not exhibit any detectable bias
across sex and ethnic subgroups and is most effective for individuals aged
between 50 and 80. Requiring only a webcam and microphone, our approach
facilitates accessible home-based PD screening, especially in regions with
limited healthcare resources.

24 Jul 2025
 National University of Singapore
National University of Singapore University College London
University College London University of Copenhagen
University of Copenhagen ETH ZürichUniversity of CologneAarhus UniversityWeizmann Institute of ScienceUniversity of RochesterKarolinska Institutet
ETH ZürichUniversity of CologneAarhus UniversityWeizmann Institute of ScienceUniversity of RochesterKarolinska Institutet University of VirginiaUniversity College DublinUniversity of Tennessee Health Science CenterUniversity of BirminghamVrije UniversiteitLoughborough UniversityUniversity of TuebingenLeiden University Medical CenterAltos LabsUniversity of Rochester Medical CenterMax Planck Institute for Biology of AgeingLongevity Escape Velocity FoundationThe Thalion InitiativeHealth Sciences North Research InstituteInstitute for Farm Animal BiologyUniversity of Oslo and Akershus University Hospital
University of VirginiaUniversity College DublinUniversity of Tennessee Health Science CenterUniversity of BirminghamVrije UniversiteitLoughborough UniversityUniversity of TuebingenLeiden University Medical CenterAltos LabsUniversity of Rochester Medical CenterMax Planck Institute for Biology of AgeingLongevity Escape Velocity FoundationThe Thalion InitiativeHealth Sciences North Research InstituteInstitute for Farm Animal BiologyUniversity of Oslo and Akershus University HospitalThe field of ageing science has gone through remarkable progress in recent decades, yet many fundamental questions remain unanswered or unexplored. Here we present a curated list of 100 open problems in ageing and longevity science. These questions were collected through community engagement and further analysed using Natural Language Processing to assess their prevalence in the literature and to identify both well-established and emerging research gaps. The final list is categorized into different topics, including molecular and cellular mechanisms of ageing, comparative biology and the use of model organisms, biomarkers, and the development of therapeutic interventions. Both long-standing questions and more recent and specific questions are featured. Our comprehensive compilation is available to the biogerontology community on our website (this http URL). Overall, this work highlights current key research questions in ageing biology and offers a roadmap for fostering future progress in biogerontology.

28 Feb 2020
We propose Artificial Intelligence Prospective Randomized Observer Blinding Evaluation (AI-PROBE) for quantitative clinical performance evaluation of radiology AI systems within prospective randomized clinical trials. AI-PROBE encompasses a study design and a matching radiology IT infrastructure that randomly blinds radiologists for results provided by AI-based image analysis. To demonstrate the applicability of our evaluation framework, we present a first prospective randomized clinical trial on the effect of Intra-Cranial Hemorrhage (ICH) detection in emergent care head CT on radiology study Turn-Around Time (TAT). Here, we acquired 620 non-contrast head CT scans from inpatient and emergency room patients at a large academic hospital. Following acquisition, scans were automatically analyzed for the presence of ICH using commercially available software (Aidoc, Tel Aviv, Israel). Cases identified positive for ICH by AI (ICH-AI+) were flagged in radiologists' reading worklists, where flagging was randomly switched off with probability 50%. TAT was measured as time difference between study completion and first clinically communicated reporting, with time stamps automatically retrieved from various IT systems. TATs for flagged cases (73+/-143 min) were significantly lower than TATs for non-flagged (132+/-193 min) cases (p<0.05, one-sided t-test), where 105 of 122 ICH-AI+ cases were true positive. Total sensitivity, specificity, and accuracy over all analyzed cases were 95.0%, 96.7%, and 96.4%, respectively. We conclude that automatic identification of ICH reduces TAT for ICH in emergent care head CT, which carries the potential for improving timely clinical management of ICH. Our results suggest that AI-PROBE can contribute to systematic quantitative evaluation of AI systems in clinical practice using clinically meaningful quantities, such as TAT or diagnostic accuracy.
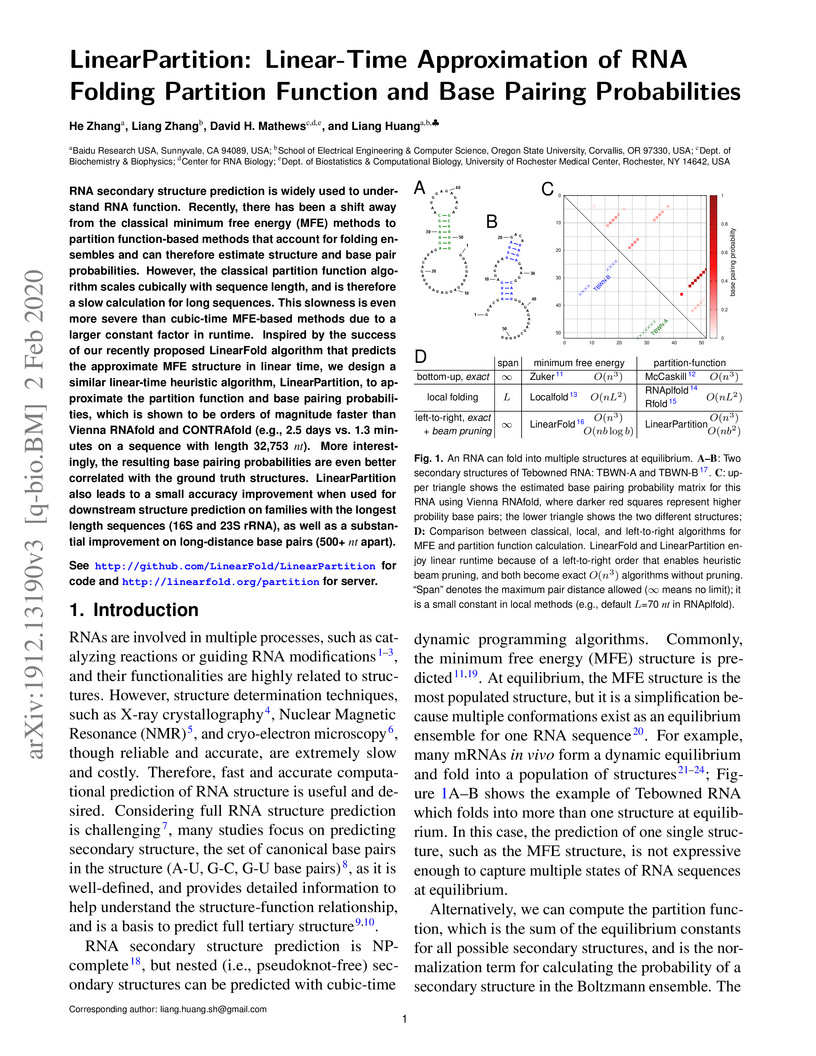
02 Feb 2020
RNA secondary structure prediction is widely used to understand RNA function.
Recently, there has been a shift away from the classical minimum free energy
(MFE) methods to partition function-based methods that account for folding
ensembles and can therefore estimate structure and base pair probabilities.
However, the classical partition function algorithm scales cubically with
sequence length, and is therefore a slow calculation for long sequences. This
slowness is even more severe than cubic-time MFE-based methods due to a larger
constant factor in runtime. Inspired by the success of our recently proposed
LinearFold algorithm that predicts the approximate MFE structure in linear
time, we design a similar linear-time heuristic algorithm, LinearPartition, to
approximate the partition function and base pairing probabilities, which is
shown to be orders of magnitude faster than Vienna RNAfold and CONTRAfold
(e.g., 2.5 days vs. 1.3 minutes on a sequence with length 32,753 nt). More
interestingly, the resulting base pairing probabilities are even better
correlated with the ground truth structures. LinearPartition also leads to a
small accuracy improvement when used for downstream structure prediction on
families with the longest length sequences (16S and 23S rRNA), as well as a
substantial improvement on long-distance base pairs (500+ nt apart).

18 Jul 2023
The classical Sankoff algorithm for the simultaneous folding and alignment of
homologous RNA sequences is highly influential, but it suffers from two major
limitations in efficiency and modeling power. First, it takes for two
sequences where n is the average sequence length. Most implementations and
variations reduce the runtime to by restricting the alignment search
space, but this is still too slow for long sequences such as full-length viral
genomes. On the other hand, the Sankoff algorithm and all its existing
implementations use a rather simplistic alignment model, which can result in
poor alignment accuracy. To address these problems, we propose LinearSankoff,
which seamlessly integrates the original Sankoff algorithm with a powerful
Hidden Markov Model-based alignment module. This extension substantially
improves alignment quality, which in turn benefits secondary structure
prediction quality, confirmed over a diverse set of RNA families. LinearSankoff
also applies beam search heuristics and the A-like algorithm to achieve
that runtime scales linearly with sequence length. LinearSankoff is the first
linear-time algorithm for simultaneous folding and alignment, and the first
such algorithm to scale to coronavirus genomes (n 30,000nt). It only
takes 10 minutes for a pair of SARS-CoV-2 and SARS-related genomes, and
outperforms previous work at identifying crucial conserved structures between
the two genomes.

04 May 2025
RNA design aims to find a sequence that folds with highest probability into a
designated target structure. However, certain structures are undesignable,
meaning no sequence can fold into the target structure under the default
(Turner) RNA folding model. Understanding the specific local structures (i.e.,
"motifs") that contribute to undesignability is crucial for refining RNA
folding models and determining the limits of RNA designability. Despite its
importance, this problem has received very little attention, and previous
efforts are neither scalable nor interpretable. We develop a new theoretical
framework for motif (un-)designability, and design scalable and interpretable
algorithms to identify minimal undesignable motifs within a given RNA secondary
structure. Our approach establishes motif undesignability by searching for
rival motifs, rather than exhaustively enumerating all (partial) sequences that
could potentially fold into the motif. Furthermore, we exploit rotational
invariance in RNA structures to detect, group, and reuse equivalent motifs and
to construct a database of unique minimal undesignable motifs. To achieve that,
we propose a loop-pair graph representation for motifs and a recursive graph
isomorphism algorithm for motif equivalence. Our algorithms successfully
identify 24 unique minimal undesignable motifs among 18 undesignable puzzles
from the Eterna100 benchmark. Surprisingly, we also find over 350 unique
minimal undesignable motifs and 663 undesignable native structures in the
ArchiveII dataset, drawn from a diverse set of RNA families. Our source code is
available at this https URL and our web server is
available at this http URL

12 Nov 2022
Although temporal coding through spike-time patterns has long been of
interest in neuroscience, the specific structures that could be useful for
spike-time codes remain highly unclear. Here, we introduce a new analytical
approach, using techniques from discrete mathematics, to study spike-time
codes. We focus on the phenomenon of ``phase precession'' in the rodent
hippocampus. During navigation and learning on a physical track, specific cells
in a rodent's brain form a highly structured pattern relative to the
oscillation of local population activity. Studies of phase precession largely
focus on its well established role in synaptic plasticity and memory formation.
Comparatively less attention has been paid to the fact that phase precession
represents one of the best candidates for a spike-time neural code. The precise
nature of this code remains an open question. Here, we derive an analytical
expression for an operator mapping points in physical space, through individual
spike times, to complex numbers. The properties of this operator highlight a
specific relationship between past and future in hippocampal spike patterns.
Importantly, this approach generalizes beyond the specific phenomenon studied
here, providing a new technique to study the neural codes within spike-time
sequences found during sensory coding and motor behavior. We then introduce a
novel spike-based decoding algorithm, based on this operator, that successfully
decodes a simulated animal's trajectory using only the animal's initial
position and a pattern of spike times. This decoder is robust to noise in spike
times and works on a timescale almost an order of magnitude shorter than
typically used with decoders that work on average firing rate. These results
illustrate the utility of a discrete approach, based on the symmetries in spike
patterns, to provide insight into the structure and function of neural systems.

17 Aug 2023
We present an artificial intelligence system to remotely assess the motor performance of individuals with Parkinson's disease (PD). Participants performed a motor task (i.e., tapping fingers) in front of a webcam, and data from 250 global participants were rated by three expert neurologists following the Movement Disorder Society Unified Parkinson's Disease Rating Scale (MDS-UPDRS). The neurologists' ratings were highly reliable, with an intra-class correlation coefficient (ICC) of 0.88. We developed computer algorithms to obtain objective measurements that align with the MDS-UPDRS guideline and are strongly correlated with the neurologists' ratings. Our machine learning model trained on these measures outperformed an MDS-UPDRS certified rater, with a mean absolute error (MAE) of 0.59 compared to the rater's MAE of 0.79. However, the model performed slightly worse than the expert neurologists (0.53 MAE). The methodology can be replicated for similar motor tasks, providing the possibility of evaluating individuals with PD and other movement disorders remotely, objectively, and in areas with limited access to neurological care.

17 Aug 2023
We present an artificial intelligence system to remotely assess the motor performance of individuals with Parkinson's disease (PD). Participants performed a motor task (i.e., tapping fingers) in front of a webcam, and data from 250 global participants were rated by three expert neurologists following the Movement Disorder Society Unified Parkinson's Disease Rating Scale (MDS-UPDRS). The neurologists' ratings were highly reliable, with an intra-class correlation coefficient (ICC) of 0.88. We developed computer algorithms to obtain objective measurements that align with the MDS-UPDRS guideline and are strongly correlated with the neurologists' ratings. Our machine learning model trained on these measures outperformed an MDS-UPDRS certified rater, with a mean absolute error (MAE) of 0.59 compared to the rater's MAE of 0.79. However, the model performed slightly worse than the expert neurologists (0.53 MAE). The methodology can be replicated for similar motor tasks, providing the possibility of evaluating individuals with PD and other movement disorders remotely, objectively, and in areas with limited access to neurological care.

30 Jan 2017

Suicide is an important but often misunderstood problem, one that researchers are now seeking to better understand through social media. Due in large part to the fuzzy nature of what constitutes suicidal risks, most supervised approaches for learning to automatically detect suicide-related activity in social media require a great deal of human labor to train. However, humans themselves have diverse or conflicting views on what constitutes suicidal thoughts. So how to obtain reliable gold standard labels is fundamentally challenging and, we hypothesize, depends largely on what is asked of the annotators and what slice of the data they label. We conducted multiple rounds of data labeling and collected annotations from crowdsourcing workers and domain experts. We aggregated the resulting labels in various ways to train a series of supervised models. Our preliminary evaluations show that using unanimously agreed labels from multiple annotators is helpful to achieve robust machine models.

11 Oct 2024
High-speed multiplex imaging of fluorescent probes is limited by a combination of spectral resolution, sensitivity, high cost and low light throughput of detectors, and filters. In this work, we present a hyperspectral detection system based on a silicon photomultiplier array that enables high-speed, high-light throughput hyperspectral imaging at low cost. We demonstrate 16 spectral channel imaging at 50 MP/s (800M spectra per second) with a conventional two photon microscope combined with a generalized spectral unmixing model that enables extraction of spectrally overlapping fluorophores. We show that the high spectral resolution combined with high throughput enables the multiplexing of multiple contrast agents over large areas and the detection of subtle spectral shifts associated with molecular binding. Silicon photomultiplier arrays may be a promising method to extend multiplex fluorescence imaging in a variety of scenarios.

09 Jan 2020
RNA structure prediction is a challenging problem, especially with
pseudoknots. Recently, there has been a shift from the classical minimum free
energy-based methods (MFE) to partition function-based ones that assemble
structures using base-pairing probabilities. Two examples of the latter group
are the popular maximum expected accuracy (MEA) method and the ProbKnot method.
ProbKnot is a fast heuristic that pairs nucleotides that are reciprocally most
probable pairing partners, and unlike MEA, can also predict structures with
pseudoknots. However, ProbKnot's full potential has been largely overlooked. In
particular, when introduced, it did not have an MEA-like hyperparameter that
can balance between positive predictive value (PPV) and sensitivity. We show
that a simple thresholded version of ProbKnot, which we call ThreshKnot, leads
to more accurate overall predictions by filtering out unlikely pairs whose
probabilities fall under a given threshold. We also show that on three
widely-used folding engines (RNAstructure, Vienna RNAfold, and CONTRAfold),
ThreshKnot always outperforms the much more involved MEA algorithm in (1) its
higher structure prediction accuracy, (2) its capability to predict
pseudoknots, and (3) its faster runtime and easier implementation. This
suggests that ThreshKnot should replace MEA as the default partition
function-based structure prediction algorithm. ThreshKnot is already available
in the widely used RNAstructure software package version 6.2 (released November
27, 2019): this https URL

13 Jul 2022
The improved diagnostic accuracy of ultrasound breast examinations remains an important goal. In this study, we propose a biophysical feature based machine learning method for breast cancer detection to improve the performance beyond a benchmark deep learning algorithm and to furthermore provide a color overlay visual map of the probability of malignancy within a lesion. This overall framework is termed disease specific imaging. Previously, 150 breast lesions were segmented and classified utilizing a modified fully convolutional network and a modified GoogLeNet, respectively. In this study multiparametric analysis was performed within the contoured lesions. Features were extracted from ultrasound radiofrequency, envelope, and log compressed data based on biophysical and morphological models. The support vector machine with a Gaussian kernel constructed a nonlinear hyperplane, and we calculated the distance between the hyperplane and data point of each feature in multiparametric space. The distance can quantitatively assess a lesion, and suggest the probability of malignancy that is color coded and overlaid onto B mode images. Training and evaluation were performed on in vivo patient data. The overall accuracy for the most common types and sizes of breast lesions in our study exceeded 98.0% for classification and 0.98 for an area under the receiver operating characteristic curve, which is more precise than the performance of radiologists and a deep learning system. Further, the correlation between the probability and BI RADS enables a quantitative guideline to predict breast cancer. Therefore, we anticipate that the proposed framework can help radiologists achieve more accurate and convenient breast cancer classification and detection.

27 Nov 2023
Pancreatic ductal adenocarcinoma (PDAC) presents a critical global health challenge, and early detection is crucial for improving the 5-year survival rate. Recent medical imaging and computational algorithm advances offer potential solutions for early diagnosis. Deep learning, particularly in the form of convolutional neural networks (CNNs), has demonstrated success in medical image analysis tasks, including classification and segmentation. However, the limited availability of clinical data for training purposes continues to provide a significant obstacle. Data augmentation, generative adversarial networks (GANs), and cross-validation are potential techniques to address this limitation and improve model performance, but effective solutions are still rare for 3D PDAC, where contrast is especially poor owing to the high heterogeneity in both tumor and background tissues. In this study, we developed a new GAN-based model, named 3DGAUnet, for generating realistic 3D CT images of PDAC tumors and pancreatic tissue, which can generate the interslice connection data that the existing 2D CT image synthesis models lack. Our innovation is to develop a 3D U-Net architecture for the generator to improve shape and texture learning for PDAC tumors and pancreatic tissue. Our approach offers a promising path to tackle the urgent requirement for creative and synergistic methods to combat PDAC. The development of this GAN-based model has the potential to alleviate data scarcity issues, elevate the quality of synthesized data, and thereby facilitate the progression of deep learning models to enhance the accuracy and early detection of PDAC tumors, which could profoundly impact patient outcomes. Furthermore, this model has the potential to be adapted to other types of solid tumors, hence making significant contributions to the field of medical imaging in terms of image processing models.

01 Mar 2024
The tasks of designing RNAs are discrete optimization problems, and several versions of these problems are NP-hard. As an alternative to commonly used local search methods, we formulate these problems as continuous optimization and develop a general framework for this optimization based on a generalization of classical partition function which we call "expected partition function". The basic idea is to start with a distribution over all possible candidate sequences, and extend the objective function from a sequence to a distribution. We then use gradient descent-based optimization methods to improve the extended objective function, and the distribution will gradually shrink towards a one-hot sequence (i.e., a single sequence). As a case study, we consider the important problem of mRNA design with wide applications in vaccines and therapeutics. While the recent work of LinearDesign can efficiently optimize mRNAs for minimum free energy (MFE), optimizing for ensemble free energy is much harder and likely intractable. Our approach can consistently improve over the LinearDesign solution in terms of ensemble free energy, with bigger improvements on longer sequences.

05 Jul 2024
Predicting the consensus structure of a set of aligned RNA homologs is a convenient method to find conserved structures in an RNA genome, which has many applications including viral diagnostics and therapeutics. However, the most commonly used tool for this task, RNAalifold, is prohibitively slow for long sequences, due to a cubic scaling with the sequence length, taking over a day on 400 SARS-CoV-2 and SARS-related genomes (~30,000nt). We present LinearAlifold, a much faster alternative that scales linearly with both the sequence length and the number of sequences, based on our work LinearFold that folds a single RNA in linear time. Our work is orders of magnitude faster than RNAalifold (0.7 hours on the above 400 genomes, or ~36 speedup) and achieves higher accuracies when compared to a database of known structures. More interestingly, LinearAlifold's prediction on SARS-CoV-2 correlates well with experimentally determined structures, substantially outperforming RNAalifold. Finally, LinearAlifold supports two energy models (Vienna and BL*) and four modes: minimum free energy (MFE), maximum expected accuracy (MEA), ThreshKnot, and stochastic sampling, each of which takes under an hour for hundreds of SARS-CoV variants. Our resource is at: this https URL (code) and this http URL (server).
There are no more papers matching your filters at the moment.
