
10 Jan 2025
The immune checkpoint inhibitors have demonstrated promising clinical efficacy across various tumor types, yet the percentage of patients who benefit from them remains low. The bindings between tumor antigens and HLA-I/TCR molecules determine the antigen presentation and T-cell activation, thereby playing an important role in the immunotherapy response. In this paper, we propose UnifyImmun, a unified cross-attention transformer model designed to simultaneously predict the bindings of peptides to both receptors, providing more comprehensive evaluation of antigen immunogenicity. We devise a two-phase strategy using virtual adversarial training that enables these two tasks to reinforce each other mutually, by compelling the encoders to extract more expressive features. Our method demonstrates superior performance in predicting both pHLA and pTCR binding on multiple independent and external test sets. Notably, on a large-scale COVID-19 pTCR binding test set without any seen peptide in training set, our method outperforms the current state-of-the-art methods by more than 10\%. The predicted binding scores significantly correlate with the immunotherapy response and clinical outcomes on two clinical cohorts. Furthermore, the cross-attention scores and integrated gradients reveal the amino-acid sites critical for peptide binding to receptors. In essence, our approach marks a significant step toward comprehensive evaluation of antigen immunogenicity.

13 Mar 2025



Commenting code is a crucial activity in software development, as it aids in
facilitating future maintenance and updates. To enhance the efficiency of
writing comments and reduce developers' workload, researchers has proposed
various automated code summarization (ACS) techniques to automatically generate
comments/summaries for given code units. However, these ACS techniques
primarily focus on generating summaries for code units at the method level.
There is a significant lack of research on summarizing higher-level code units,
such as file-level and module-level code units, despite the fact that summaries
of these higher-level code units are highly useful for quickly gaining a
macro-level understanding of software components and architecture. To fill this
gap, in this paper, we conduct a systematic study on how to use LLMs for
commenting higher-level code units, including file level and module level.
These higher-level units are significantly larger than method-level ones, which
poses challenges in handling long code inputs within LLM constraints and
maintaining efficiency. To address these issues, we explore various
summarization strategies for ACS of higher-level code units, which can be
divided into three types: full code summarization, reduced code summarization,
and hierarchical code summarization. The experimental results suggest that for
summarizing file-level code units, using the full code is the most effective
approach, with reduced code serving as a cost-efficient alternative. However,
for summarizing module-level code units, hierarchical code summarization
becomes the most promising strategy. In addition, inspired by the research on
method-level ACS, we also investigate using the LLM as an evaluator to evaluate
the quality of summaries of higher-level code units. The experimental results
demonstrate that the LLM's evaluation results strongly correlate with human
evaluations.

05 Aug 2025
Polyphenols and proteins are essential biomolecules that influence food functionality and, by extension, human health. Their interactions -- hereafter referred to as PhPIs (polyphenol-protein interactions) -- affect key processes such as nutrient bioavailability, antioxidant activity, and therapeutic efficacy. However, these interactions remain challenging due to the structural diversity of polyphenols and the dynamic nature of protein binding. Traditional experimental techniques like nuclear magnetic resonance (NMR) and mass spectrometry (MS), along with computational tools such as molecular docking and molecular dynamics (MD), have offered important insights but face constraints in scalability, throughput, and reproducibility. This review explores how deep learning (DL) is reshaping the study of PhPIs by enabling efficient prediction of binding sites, interaction affinities, and MD using high-dimensional bio- and chem-informatics data. While DL enhances prediction accuracy and reduces experimental redundancy, its effectiveness remains limited by data availability, quality, and representativeness, particularly in the context of natural products. We critically assess current DL frameworks for PhPIs analysis and outline future directions, including multimodal data integration, improved model generalizability, and development of domain-specific benchmark datasets. This synthesis offers guidance for researchers aiming to apply DL in unraveling structure-function relationships of polyphenols, accelerating discovery in nutritional science and therapeutic development.

03 Apr 2025
The output prediction of quantum circuits is a formidably challenging task
imperative in developing quantum devices. Motivated by the natural graph
representation of quantum circuits, this paper proposes a Graph Neural Networks
(GNNs)-based framework to predict the output expectation values of quantum
circuits under noisy and noiseless conditions and compare the performance of
different parameterized quantum circuits (PQCs). We construct datasets under
noisy and noiseless conditions using a non-parameterized quantum gate set to
predict circuit expectation values. The node feature vectors for GNNs are
specifically designed to include noise information. In our simulations, we
compare the prediction performance of GNNs in both noisy and noiseless
conditions against Convolutional Neural Networks (CNNs) on the same dataset and
their qubit scalability. GNNs demonstrate superior prediction accuracy across
diverse conditions. Subsequently, we utilize the parameterized quantum gate set
to construct noisy PQCs and compute the ground state energy of hydrogen
molecules using the Variational Quantum Eigensolver (VQE). We propose two
schemes: the Indirect Comparison scheme, which involves directly predicting the
ground state energy and subsequently comparing circuit performances, and the
Direct Comparison scheme, which directly predicts the relative performance of
the two circuits. Simulation results indicate that the Direct Comparison scheme
significantly outperforms the Indirect Comparison scheme by an average of 36.2%
on the same dataset, providing a new and effective perspective for using GNNs
to predict the overall properties of PQCs, specifically by focusing on their
performance differences.

12 Jul 2025

Colorectal cancer (CRC) is the third most diagnosed cancer and the second leading cause of cancer-related death worldwide. Accurate histopathological grading of CRC is essential for prognosis and treatment planning but remains a subjective process prone to observer variability and limited by global shortages of trained pathologists. To promote automated and standardized solutions, we organized the ICIP Grand Challenge on Colorectal Cancer Tumor Grading and Segmentation using the publicly available METU CCTGS dataset. The dataset comprises 103 whole-slide images with expert pixel-level annotations for five tissue classes. Participants submitted segmentation masks via Codalab, evaluated using metrics such as macro F-score and mIoU. Among 39 participating teams, six outperformed the Swin Transformer baseline (62.92 F-score). This paper presents an overview of the challenge, dataset, and the top-performing methods

09 Sep 2024
Predictive learning for spatio-temporal processes (PL-STP) on complex spatial domains plays a critical role in various scientific and engineering fields, with its essence being the construction of operators between infinite-dimensional function spaces. This paper focuses on the unequal-domain mappings in PL-STP and categorising them into increase-domain and decrease-domain mapping. Recent advances in deep learning have revealed the great potential of neural operators (NOs) to learn operators directly from observational data. However, existing NOs require input space and output space to be the same domain, which pose challenges in ensuring predictive accuracy and stability for unequal-domain mappings. To this end, this study presents a general reduced-order neural operator named Reduced-Order Neural Operator on Riemannian Manifolds (RO-NORM), which consists of two parts: the unequal-domain encoder/decoder and the same-domain approximator. Motivated by the variable separation in classical modal decomposition, the unequal-domain encoder/decoder uses the pre-computed bases to reformulate the spatio-temporal function as a sum of products between spatial (or temporal) bases and corresponding temporally (or spatially) distributed weight functions, thus the original unequal-domain mapping can be converted into a same-domain mapping. Consequently, the same-domain approximator NORM is applied to model the transformed mapping. The performance of our proposed method has been evaluated on six benchmark cases, including parametric PDEs, engineering and biomedical applications, and compared with four baseline algorithms: DeepONet, POD-DeepONet, PCA-Net, and vanilla NORM. The experimental results demonstrate the superiority of RO-NORM in prediction accuracy and training efficiency for PL-STP.

05 Aug 2025

Mixed precision quantization (MPQ) is an effective quantization approach to achieve accuracy-complexity trade-off of neural network, through assigning different bit-widths to network activations and weights in each layer. The typical way of existing MPQ methods is to optimize quantization policies (i.e., bit-width allocation) in a gradient descent manner, termed as Differentiable (DMPQ). At the end of the search, the bit-width associated to the quantization parameters which has the largest value will be selected to form the final mixed precision quantization policy, with the implicit assumption that the values of quantization parameters reflect the operation contribution to the accuracy improvement. While much has been discussed about the MPQ improvement, the bit-width selection process has received little attention. We study this problem and argue that the magnitude of quantization parameters does not necessarily reflect the actual contribution of the bit-width to the task performance. Then, we propose a Shapley-based MPQ (SMPQ) method, which measures the bit-width operation direct contribution on the MPQ task. To reduce computation cost, a Monte Carlo sampling-based approximation strategy is proposed for Shapley computation. Extensive experiments on mainstream benchmarks demonstrate that our SMPQ consistently achieves state-of-the-art performance than gradient-based competitors.
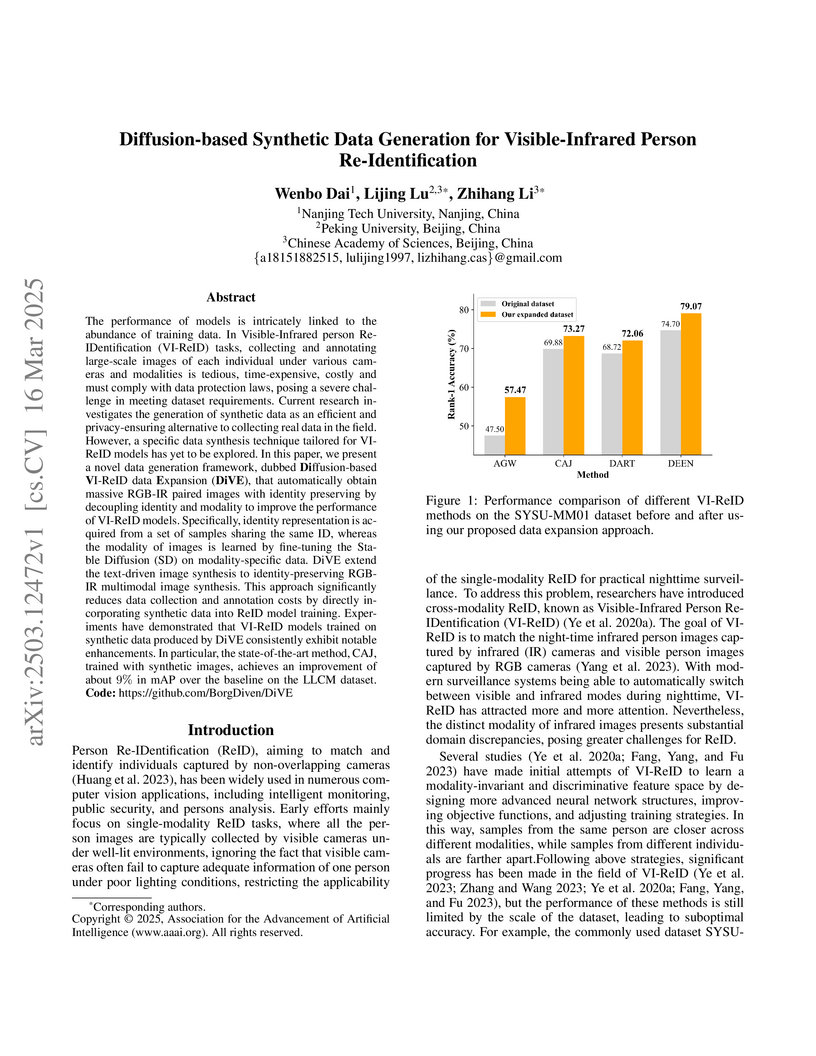
16 Mar 2025

DiVE, a framework developed by researchers at Nanjing Tech University, Peking University, and the Chinese Academy of Sciences, generates identity-preserving synthetic visible-infrared person images by leveraging a pre-trained diffusion model to decouple identity from modality information, thereby enhancing the performance of various state-of-the-art VI-ReID models.

20 Dec 2024

Current superconducting quantum devices impose strict connectivity
constraints on quantum circuit execution, necessitating circuit transformation
before executing quantum circuits on physical hardware. Numerous quantum
circuit transformation (QCT) algorithms have been proposed. To enable faithful
evaluation of state-of-the-art QCT algorithms, this paper introduces QKNOB
(Qubit mapping Benchmark with Known Near-Optimality), a novel benchmark
construction method for QCT. QKNOB circuits have built-in transformations with
near-optimal (close to the theoretical optimum) SWAP count and depth overhead.
QKNOB provides general and unbiased evaluation of QCT algorithms. Using QKNOB,
we demonstrate that SABRE, the default Qiskit compiler, consistently achieves
the best performance on the 53-qubit IBM Q Rochester and Google Sycamore
devices for both SWAP count and depth objectives. Our results also reveal
significant performance gaps relative to the near-optimal transformation costs
of QKNOB. Our construction algorithm and benchmarks are open-source.

06 Jul 2021
DeepDDS: deep graph neural network with attention mechanism to predict synergistic drug combinations
DeepDDS: deep graph neural network with attention mechanism to predict synergistic drug combinations
Drug combination therapy has become a increasingly promising method in the
treatment of cancer. However, the number of possible drug combinations is so
huge that it is hard to screen synergistic drug combinations through wet-lab
experiments. Therefore, computational screening has become an important way to
prioritize drug combinations. Graph neural network have recently shown
remarkable performance in the prediction of compound-protein interactions, but
it has not been applied to the screening of drug combinations. In this paper,
we proposed a deep learning model based on graph neural networks and attention
mechanism to identify drug combinations that can effectively inhibit the
viability of specific cancer cells. The feature embeddings of drug molecule
structure and gene expression profiles were taken as input to multi-layer
feedforward neural network to identify the synergistic drug combinations. We
compared DeepDDS with classical machine learning methods and other deep
learning-based methods on benchmark data set, and the leave-one-out
experimental results showed that DeepDDS achieved better performance than
competitive methods. Also, on an independent test set released by well-known
pharmaceutical enterprise AstraZeneca, DeepDDS was superior to competitive
methods by more than 16\% predictive precision. Furthermore, we explored the
interpretability of the graph attention network, and found the correlation
matrix of atomic features revealed important chemical substructures of drugs.
We believed that DeepDDS is an effective tool that prioritized synergistic drug
combinations for further wet-lab experiment validation.

04 Dec 2024
The anti-cancer immune response relies on the bindings between T-cell
receptors (TCRs) and antigens, which elicits adaptive immunity to eliminate
tumor cells. This ability of the immune system to respond to novel various
neoantigens arises from the immense diversity of TCR repository. However, TCR
diversity poses a significant challenge on accurately predicting antigen-TCR
bindings. In this study, we introduce a lightweight masked language model,
termed tcrLM, to address this challenge. Our approach involves randomly masking
segments of TCR sequences and training tcrLM to infer the masked segments,
thereby enabling the extraction of expressive features from TCR sequences. To
further enhance robustness, we incorporate virtual adversarial training into
tcrLM. We construct the largest TCR CDR3 sequence set with more than 100
million distinct sequences, and pretrain tcrLM on these sequences. The
pre-trained encoder is subsequently applied to predict TCR-antigen binding
specificity. We evaluate model performance on three test datasets: independent,
external, and COVID-19 test set. The results demonstrate that tcrLM not only
surpasses existing TCR-antigen binding prediction methods, but also outperforms
other mainstream protein language models. More interestingly, tcrLM effectively
captures the biochemical properties and positional preference of amino acids
within TCR sequences. Additionally, the predicted TCR-neoantigen binding scores
indicates the immunotherapy responses and clinical outcomes in a melanoma
cohort. These findings demonstrate the potential of tcrLM in predicting
TCR-antigen binding specificity, with significant implications for advancing
immunotherapy and personalized medicine.

15 Nov 2021
During the curing process of composites, the temperature history heavily determines the evolutions of the field of degree of cure as well as the residual stress, which will further influence the mechanical properties of composite, thus it is important to simulate the real temperature history to optimize the curing process of composites. Since thermochemical analysis using Finite Element (FE) simulations requires heavy computational loads and data-driven approaches suffer from the complexity of highdimensional mapping. This paper proposes a Residual Fourier Neural Operator (ResFNO) to establish the direct high-dimensional mapping from any given cure cycle to the corresponding temperature histories. By integrating domain knowledge into a time-resolution independent parameterized neural network, the mapping between cure cycles to temperature histories can be learned using limited number of labelled data. Besides, a novel Fourier residual mapping is designed based on mode decomposition to accelerate the training and boost the performance significantly. Several cases are carried out to evaluate the superior performance and generalizability of the proposed method comprehensively.

01 Oct 2025
Let be a ring with identity and be its Jacobson radical. Assume that is -invertible and . This paper provides necessary and sufficient conditions for to be -invertible. As an application, corresponding results on generalized inverses of dual matrices are derived.

28 May 2024

We introduce an efficient method for learning linear models from uncertain data, where uncertainty is represented as a set of possible variations in the data, leading to predictive multiplicity. Our approach leverages abstract interpretation and zonotopes, a type of convex polytope, to compactly represent these dataset variations, enabling the symbolic execution of gradient descent on all possible worlds simultaneously. We develop techniques to ensure that this process converges to a fixed point and derive closed-form solutions for this fixed point. Our method provides sound over-approximations of all possible optimal models and viable prediction ranges. We demonstrate the effectiveness of our approach through theoretical and empirical analysis, highlighting its potential to reason about model and prediction uncertainty due to data quality issues in training data.

07 Sep 2024
The qubit mapping problem (QMP) focuses on the mapping and routing of qubits in quantum circuits so that the strict connectivity constraints imposed by near-term quantum hardware are satisfied. QMP is a pivotal task for quantum circuit compilation and its decision version is NP-complete. In this study, we present an effective approach called Adaptive Divided-And-Conqure (ADAC) to solve QMP. Our ADAC algorithm adaptively partitions circuits by leveraging subgraph isomorphism and ensuring coherence among subcircuits. Additionally, we employ a heuristic approach to optimise the routing algorithm during circuit partitioning. Through extensive experiments across various NISQ devices and circuit benchmarks, we demonstrate that the proposed ADAC algorithm outperforms the state-of-the-art method. Specifically, ADAC shows an improvement of nearly 50\% on the IBM Tokyo architecture. Furthermore, ADAC exhibits an improvement of around 18\% on pseudo-realistic circuits implemented on grid-like architectures with larger qubit numbers, where the pseudo-realistic circuits are constructed based on the characteristics of widely existing realistic circuits, aiming to investigate the applicability of ADAC. Our findings highlight the potential of ADAC in quantum circuit compilation and the deployment of practical applications on near-term quantum hardware platforms.

05 Jul 2025
The equation of state (EoS) for neutron stars is a crucial topic in astrophysics, nuclear physics, and quantum chromodynamics (QCD), influencing their structure, stability, and observable properties. This review classifies EoS models into hadronic matter, hybrid, and quark matter models, analyzing their assumptions, predictions, and constraints. While hadronic models characterize nucleonic matter, potentially including contributions from hyperons or mesons, hybrid models introduce phase transitions to quark matter, and quark models hypothesize the presence of deconfined quark matter cores or entirely quark-composed stars. By synthesizing results from recent theoretical and observational studies, this review aims to offer a comprehensive understanding of the methodologies used in constructing neutron star EoS, their implications, and future directions.

07 Jan 2025
The advancement of single-cell sequencing technology has promoted the
generation of a large amount of single-cell transcriptional profiles, providing
unprecedented opportunities to identify drug-resistant cell subpopulations
within a tumor. However, few studies have focused on drug response prediction
at single-cell level, and their performance remains suboptimal. This paper
proposed scAdaDrug, a novel multi-source domain adaptation model powered by
adaptive importance-aware representation learning to predict drug response of
individual cells. We used a shared encoder to extract domain-invariant features
related to drug response from multiple source domains by utilizing adversarial
domain adaptation. Particularly, we introduced a plug-and-play module to
generate importance-aware and mutually independent weights, which could
adaptively modulate the latent representation of each sample in element-wise
manner between source and target domains. Extensive experimental results showed
that our model achieved state-of-the-art performance in predicting drug
response on multiple independent datasets, including single-cell datasets
derived from both cell lines and patient-derived xenografts (PDX) models, as
well as clinical tumor patient cohorts. Moreover, the ablation experiments
demonstrated our model effectively captured the underlying patterns determining
drug response from multiple source domains.

30 Jul 2022
Attributed graph clustering is one of the most fundamental tasks among graph
learning field, the goal of which is to group nodes with similar
representations into the same cluster without human annotations. Recent studies
based on graph contrastive learning method have achieved remarkable results
when exploit graph-structured data. However, most existing methods 1) do not
directly address the clustering task, since the representation learning and
clustering process are separated; 2) depend too much on data augmentation,
which greatly limits the capability of contrastive learning; 3) ignore the
contrastive message for clustering tasks, which adversely degenerate the
clustering results. In this paper, we propose a Neighborhood Contrast Framework
for Attributed Graph Clustering, namely NCAGC, seeking for conquering the
aforementioned limitations. Specifically, by leveraging the Neighborhood
Contrast Module, the representation of neighbor nodes will be 'push closer' and
become clustering-oriented with the neighborhood contrast loss. Moreover, a
Contrastive Self-Expression Module is built by minimizing the node
representation before and after the self-expression layer to constraint the
learning of self-expression matrix. All the modules of NCAGC are optimized in a
unified framework, so the learned node representation contains
clustering-oriented messages. Extensive experimental results on four attributed
graph datasets demonstrate the promising performance of NCAGC compared with 16
state-of-the-art clustering methods. The code is available at
this https URL

20 Jun 2024
The Diffeomorphism Neural Operator (DNO) learns solutions to partial differential equations across varying geometric domains and physical parameters by mapping diverse domains to a generic domain. It demonstrates robust generalization, achieving L2 errors consistently below 0.05 on unseen domain shapes and scales for various 2D and 3D PDE problems, including complex fluid dynamics and machining deformation.

26 Jul 2022
Target localization based on frequency diverse array (FDA) radar has lately garnered significant research interest. A linear frequency offset (FO) across FDA antennas yields a range-angle dependent beampattern that allows for joint estimation of range and direction-of-arrival (DoA). Prior works on FDA largely focus on the one-dimensional linear array to estimate only azimuth angle and range while ignoring the elevation and Doppler velocity. However, in many applications, the latter two parameters are also essential for target localization. Further, there is also an interest in radar systems that employ fewer measurements in temporal, Doppler, or spatial signal domains. We address these multiple challenges by proposing a co-prime L-shaped FDA, wherein co-prime FOs are applied across the elements of L-shaped co-prime array and each element transmits at a non-uniform co-prime pulse repetition interval (C or C-Cube). This co-pulsing FDA yields significantly large degrees-of-freedom (DoFs) for target localization in the range-azimuth-elevation-Doppler domain while also reducing the time-on-target and transmit spectral usage. By exploiting these DoFs, we develop C-Cube auto-pairing (CCing) algorithm, in which all the parameters are ipso facto paired during a joint estimation. We show that C-Cube FDA requires at least antenna elements and pulses to guarantee perfect recovery of targets as against elements and pulses required by both L-shaped uniform linear array and L-shaped linear FO FDA with uniform pulsing. We derive Cramér-Rao bounds (CRBs) for joint angle-range-Doppler estimation errors for C-Cube FDA and provide the conditions under which the CRBs exist. Numerical experiments with our CCing algorithm show great performance improvements in parameter recovery.
There are no more papers matching your filters at the moment.
