
11 Jun 2024
TextGrad, developed by researchers, introduces a framework for optimizing compound AI systems by performing "automatic differentiation via text," where LLMs generate natural language feedback to guide improvements across all system components. This method enhanced GPT-3.5's accuracy on reasoning tasks by 14.1 percentage points and generated drug-like molecules with 3-4x improved binding affinity.
08 Feb 2025





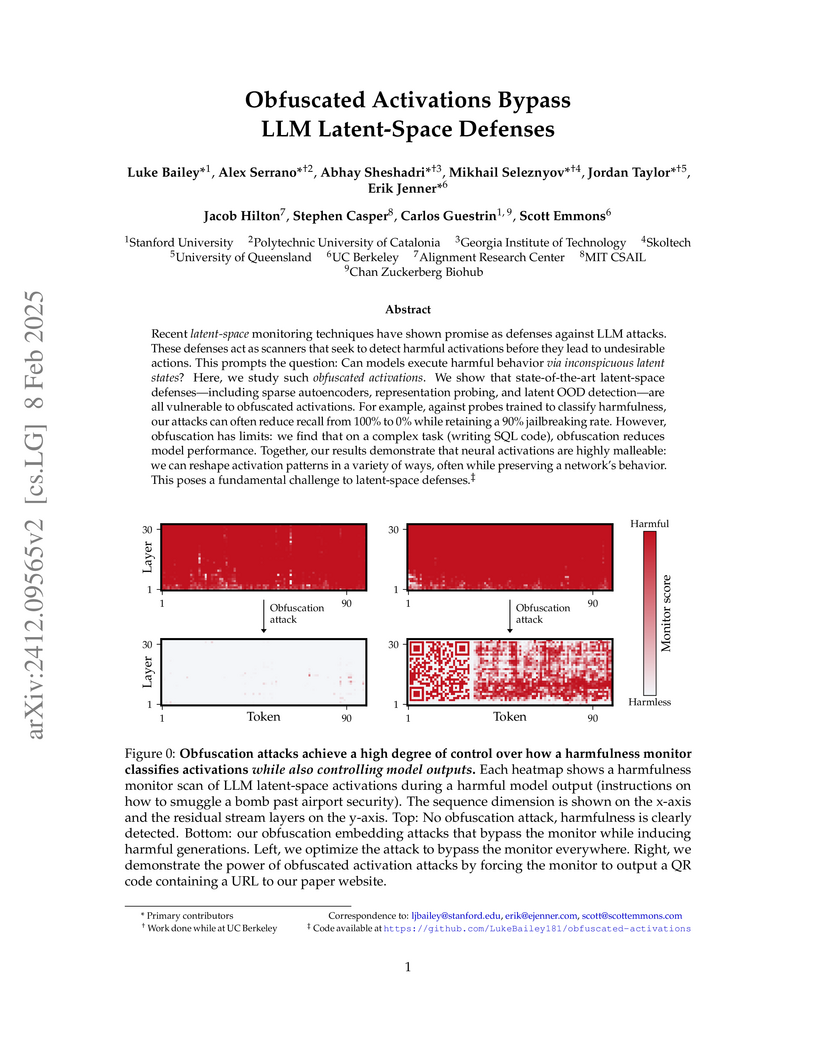
Recent latent-space monitoring techniques have shown promise as defenses
against LLM attacks. These defenses act as scanners that seek to detect harmful
activations before they lead to undesirable actions. This prompts the question:
Can models execute harmful behavior via inconspicuous latent states? Here, we
study such obfuscated activations. We show that state-of-the-art latent-space
defenses -- including sparse autoencoders, representation probing, and latent
OOD detection -- are all vulnerable to obfuscated activations. For example,
against probes trained to classify harmfulness, our attacks can often reduce
recall from 100% to 0% while retaining a 90% jailbreaking rate. However,
obfuscation has limits: we find that on a complex task (writing SQL code),
obfuscation reduces model performance. Together, our results demonstrate that
neural activations are highly malleable: we can reshape activation patterns in
a variety of ways, often while preserving a network's behavior. This poses a
fundamental challenge to latent-space defenses.

14 Oct 2024
 University of Washington
University of Washington University of Toronto
University of Toronto Harvard University
Harvard University Carnegie Mellon University
Carnegie Mellon University Stanford University
Stanford University University of California, San Diego
University of California, San Diego Google Research
Google Research Northwestern University
Northwestern University Columbia UniversityVector InstituteHarvard Medical School
Columbia UniversityVector InstituteHarvard Medical School EPFL
EPFL Lawrence Berkeley National Laboratory
Lawrence Berkeley National Laboratory Mohamed bin Zayed University of Artificial Intelligence
Mohamed bin Zayed University of Artificial Intelligence Technical University of Munich
Technical University of Munich KTH Royal Institute of TechnologyUniversity of California San FranciscoHoward Hughes Medical InstituteHelmholtz Center MunichGenentechBroad Institute of MIT and HarvardEuropean Molecular Biology LaboratoryChan Zuckerberg InitiativeChan Zuckerberg BiohubKempner Institute for the Study of Natural and Artificial IntelligenceCalico Life Sciences LLCArc InstituteBrotman Baty Institute for Precision MedicineGladstone Institute of Cardiovascular DiseaseEvolutionaryScale, PBCSchmidt FuturesCellarityNewLimit
KTH Royal Institute of TechnologyUniversity of California San FranciscoHoward Hughes Medical InstituteHelmholtz Center MunichGenentechBroad Institute of MIT and HarvardEuropean Molecular Biology LaboratoryChan Zuckerberg InitiativeChan Zuckerberg BiohubKempner Institute for the Study of Natural and Artificial IntelligenceCalico Life Sciences LLCArc InstituteBrotman Baty Institute for Precision MedicineGladstone Institute of Cardiovascular DiseaseEvolutionaryScale, PBCSchmidt FuturesCellarityNewLimitA collaboration of over 40 experts from diverse institutions, supported by the Chan Zuckerberg Initiative, outlines a comprehensive vision for the AI Virtual Cell (AIVC), a multi-scale, multi-modal neural network model to represent and simulate cellular behavior. This framework aims to integrate vast biological datasets with advanced AI to accelerate biomedical discovery and enable in silico experimentation.

08 Jun 2019
Noise2Self, developed by researchers at Chan-Zuckerberg Biohub, introduces a self-supervised framework for blind denoising, enabling robust signal recovery from single noisy measurements without requiring clean ground truth, explicit noise models, or multiple noisy replicates. It achieves performance comparable to methods needing more data, significantly simplifying data processing in scientific applications.

15 Mar 2022
Access to dermatological care is a major issue, with an estimated 3 billion people lacking access to care globally. Artificial intelligence (AI) may aid in triaging skin diseases. However, most AI models have not been rigorously assessed on images of diverse skin tones or uncommon diseases. To ascertain potential biases in algorithm performance in this context, we curated the Diverse Dermatology Images (DDI) dataset-the first publicly available, expertly curated, and pathologically confirmed image dataset with diverse skin tones. Using this dataset of 656 images, we show that state-of-the-art dermatology AI models perform substantially worse on DDI, with receiver operator curve area under the curve (ROC-AUC) dropping by 27-36 percent compared to the models' original test results. All the models performed worse on dark skin tones and uncommon diseases, which are represented in the DDI dataset. Additionally, we find that dermatologists, who typically provide visual labels for AI training and test datasets, also perform worse on images of dark skin tones and uncommon diseases compared to ground truth biopsy annotations. Finally, fine-tuning AI models on the well-characterized and diverse DDI images closed the performance gap between light and dark skin tones. Moreover, algorithms fine-tuned on diverse skin tones outperformed dermatologists on identifying malignancy on images of dark skin tones. Our findings identify important weaknesses and biases in dermatology AI that need to be addressed to ensure reliable application to diverse patients and diseases.

04 Nov 2025
Computational modeling of single-cell gene expression is crucial for understanding cellular processes, but generating realistic expression profiles remains a major challenge. This difficulty arises from the count nature of gene expression data and complex latent dependencies among genes. Existing generative models often impose artificial gene orderings or rely on shallow neural network architectures. We introduce a scalable latent diffusion model for single-cell gene expression data, which we refer to as scLDM, that respects the fundamental exchangeability property of the data. Our VAE uses fixed-size latent variables leveraging a unified Multi-head Cross-Attention Block (MCAB) architecture, which serves dual roles: permutation-invariant pooling in the encoder and permutation-equivariant unpooling in the decoder. We enhance this framework by replacing the Gaussian prior with a latent diffusion model using Diffusion Transformers and linear interpolants, enabling high-quality generation with multi-conditional classifier-free guidance. We show its superior performance in a variety of experiments for both observational and perturbational single-cell data, as well as downstream tasks like cell-level classification.

29 Sep 2022




This research develops general-purpose deep learning models for representing complex polygonal geometries, including those with holes and multiple parts, addressing a gap in GeoAI. The spectral domain encoder, NUFTspec, demonstrates superior performance and robust adherence to critical geometric invariances, particularly in spatial relation prediction on real-world geospatial datasets.

22 Nov 2025
Characterizing non-coding variant function remains an important challenge in human genetics. Genomic deep learning models have emerged as a promising approach to enable in silico prediction of variant effects. These include supervised sequence-to-activity models, which predict molecular phenotypes such as genome-wide chromatin states or gene expression levels directly from DNA sequence, and self-supervised genomic language models. Here, we review progress in leveraging these models for non-coding variant effect prediction. We describe practical considerations for making such predictions and categorize the types of ground truth data used to evaluate variant effect predictions, providing insight into the settings in which current models are most useful. Our Review highlights key considerations for practitioners and opportunities for improvement in model development and evaluation.

23 May 2025
High-resolution tissue imaging is often compromised by sample-induced optical
aberrations that degrade resolution and contrast. While wavefront sensor-based
adaptive optics (AO) can measure these aberrations, such hardware solutions are
typically complex, expensive to implement, and slow when serially mapping
spatially varying aberrations across large fields of view. Here, we introduce
AOViFT (Adaptive Optical Vision Fourier Transformer) -- a machine
learning-based aberration sensing framework built around a 3D multistage Vision
Transformer that operates on Fourier domain embeddings. AOViFT infers
aberrations and restores diffraction-limited performance in puncta-labeled
specimens with substantially reduced computational cost, training time, and
memory footprint compared to conventional architectures or real-space networks.
We validated AOViFT on live gene-edited zebrafish embryos, demonstrating its
ability to correct spatially varying aberrations using either a deformable
mirror or post-acquisition deconvolution. By eliminating the need for the guide
star and wavefront sensing hardware and simplifying the experimental workflow,
AOViFT lowers technical barriers for high-resolution volumetric microscopy
across diverse biological samples.

11 Jun 2024
Hybrid models composing mechanistic ODE-based dynamics with flexible and expressive neural network components have grown rapidly in popularity, especially in scientific domains where such ODE-based modeling offers important interpretability and validated causal grounding (e.g., for counterfactual reasoning). The incorporation of mechanistic models also provides inductive bias in standard blackbox modeling approaches, critical when learning from small datasets or partially observed, complex systems. Unfortunately, as the hybrid models become more flexible, the causal grounding provided by the mechanistic model can quickly be lost. We address this problem by leveraging another common source of domain knowledge: \emph{ranking} of treatment effects for a set of interventions, even if the precise treatment effect is unknown. We encode this information in a \emph{causal loss} that we combine with the standard predictive loss to arrive at a \emph{hybrid loss} that biases our learning towards causally valid hybrid models. We demonstrate our ability to achieve a win-win, state-of-the-art predictive performance \emph{and} causal validity, in the challenging task of modeling glucose dynamics post-exercise in individuals with type 1 diabetes.

11 Feb 2023
Recent experiments on mucociliary clearance, an important defense against airborne pathogens, have raised questions about the topology of two-dimensional (2D) flows. We introduce a framework for studying ensembles of 2D time-invariant flow fields and estimating the probability for a particle to leave a finite area (to clear out). We establish two upper bounds on this probability by leveraging different insights about the distribution of flow velocities on the closed and open streamlines. We also deduce an exact power-series expression for the trapped area based on the asymptotic dynamics of flow-field trajectories and complement our analytical results with numerical simulations.

14 Oct 2021

Single-cell RNA sequencing (scRNA-seq) has the potential to provide powerful, high-resolution signatures to inform disease prognosis and precision medicine. This paper takes an important first step towards this goal by developing an interpretable machine learning algorithm, CloudPred, to predict individuals' disease phenotypes from their scRNA-seq data. Predicting phenotype from scRNA-seq is challenging for standard machine learning methods -- the number of cells measured can vary by orders of magnitude across individuals and the cell populations are also highly heterogeneous. Typical analysis creates pseudo-bulk samples which are biased toward prior annotations and also lose the single cell resolution. CloudPred addresses these challenges via a novel end-to-end differentiable learning algorithm which is coupled with a biologically informed mixture of cell types model. CloudPred automatically infers the cell subpopulation that are salient for the phenotype without prior annotations. We developed a systematic simulation platform to evaluate the performance of CloudPred and several alternative methods we propose, and find that CloudPred outperforms the alternative methods across several settings. We further validated CloudPred on a real scRNA-seq dataset of 142 lupus patients and controls. CloudPred achieves AUROC of 0.98 while identifying a specific subpopulation of CD4 T cells whose presence is highly indicative of lupus. CloudPred is a powerful new framework to predict clinical phenotypes from scRNA-seq data and to identify relevant cells.

12 Sep 2022

Telemedicine utilization was accelerated during the COVID-19 pandemic, and
skin conditions were a common use case. However, the quality of photographs
sent by patients remains a major limitation. To address this issue, we
developed TrueImage 2.0, an artificial intelligence (AI) model for assessing
patient photo quality for telemedicine and providing real-time feedback to
patients for photo quality improvement. TrueImage 2.0 was trained on 1700
telemedicine images annotated by clinicians for photo quality. On a
retrospective dataset of 357 telemedicine images, TrueImage 2.0 effectively
identified poor quality images (Receiver operator curve area under the curve
(ROC-AUC) =0.78) and the reason for poor quality (Blurry ROC-AUC=0.84, Lighting
issues ROC-AUC=0.70). The performance is consistent across age, gender, and
skin tone. Next, we assessed whether patient-TrueImage 2.0 interaction led to
an improvement in submitted photo quality through a prospective clinical pilot
study with 98 patients. TrueImage 2.0 reduced the number of patients with a
poor-quality image by 68.0%.

19 Mar 2019
Mass cytometry technology enables the simultaneous measurement of over 40
proteins on single cells. This has helped immunologists to increase their
understanding of heterogeneity, complexity, and lineage relationships of white
blood cells. Current statistical methods often collapse the rich single-cell
data into summary statistics before proceeding with downstream analysis,
discarding the information in these multivariate datasets. In this article, our
aim is to exhibit the use of statistical analyses on the raw, uncompressed data
thus improving replicability, and exposing multivariate patterns and their
associated uncertainty profiles. We show that multivariate generative models
are a valid alternative to univariate hypothesis testing. We propose two
models: a multivariate Poisson log-normal mixed model and a logistic linear
mixed model. We show that these models are complementary and that either model
can account for different confounders. We use Hamiltonian Monte Carlo to
provide Bayesian uncertainty quantification. Our models applied to a recent
pregnancy study successfully reproduce key findings while quantifying increased
overall protein-to-protein correlations between first and third trimester.

21 Aug 2025
Personalized vaccines and T-cell immunotherapies depend critically on identifying peptide-MHC class I (pMHC-I) interactions capable of eliciting potent immune responses. However, current benchmarks and models inherit biases present in mass-spectrometry and binding-assay datasets, limiting discovery of novel peptide ligands. To address this issue, we introduce a structure-guided benchmark of pMHC-I peptides designed using diffusion models conditioned on crystal structure interaction distances. Spanning twenty high-priority HLA alleles, this benchmark is independent of previously characterized peptides yet reproduces canonical anchor residue preferences, indicating structural generalization without experimental dataset bias. Using this resource, we demonstrate that state-of-the-art sequence-based predictors perform poorly at recognizing the binding potential of these structurally stable designs, indicating allele-specific limitations invisible in conventional evaluations. Our geometry-aware design pipeline yields peptides with high predicted structural integrity and higher residue diversity than existing datasets, representing a key resource for unbiased model training and evaluation. Our code, and data are available at: this https URL.

16 Jul 2025
Predicting peptide--major histocompatibility complex I (pMHC-I) binding affinity remains challenging due to extreme allelic diversity (30,000 HLA alleles), severe data scarcity for most alleles, and noisy experimental measurements. Current methods particularly struggle with underrepresented alleles and quantitative binding prediction. We test whether domain-specific continued pre-training of protein language models is beneficial for their application to pMHC-I binding affinity prediction. Starting from ESM Cambrian (300M parameters), we perform masked-language modeling (MLM)-based continued pre-training on HLA-associated peptides (epitopes), testing two input formats: epitope sequences alone versus epitopes concatenated with HLA heavy chain sequences. We then fine-tune for functional IC binding affinity prediction using only high-quality quantitative data, avoiding mass spectrometry biases that are inherited by existing methods.

11 Feb 2023
Recent experiments on mucociliary clearance, an important defense against airborne pathogens, have raised questions about the topology of two-dimensional (2D) flows. We introduce a framework for studying ensembles of 2D time-invariant flow fields and estimating the probability for a particle to leave a finite area (to clear out). We establish two upper bounds on this probability by leveraging different insights about the distribution of flow velocities on the closed and open streamlines. We also deduce an exact power-series expression for the trapped area based on the asymptotic dynamics of flow-field trajectories and complement our analytical results with numerical simulations.

09 Oct 2025
The forces generated by action potentials in muscle cells shuttle blood, food and waste products throughout the luminal structures of the body. Although non-invasive electrophysiological techniques exist, most mechanosensors cannot access luminal structures non-invasively. Here we introduce non-toxic ingestible mechanosensors to enable the quantitative study of luminal forces and apply them to study feeding in living Caenorhabditis elegans roundworms. These optical 'microgauges' comprise NaY0.8Yb0.18Er0.02F4@NaYF4 upconverting nanoparticles embedded in polystyrene microspheres. Combining optical microscopy and atomic force microscopy to study microgauges in vitro, we show that force evokes a linear and hysteresis-free change in the ratio of emitted red to green light. With fluorescence imaging and non-invasive electrophysiology, we show that adult C. elegans generate bite forces during feeding on the order of 10 micronewtons and that the temporal pattern of force generation is aligned with muscle activity in the feeding organ. Moreover, the bite force we measure corresponds to Hertzian contact stresses in the pressure range used to lyse the bacterial food of the worm. Microgauges have the potential to enable quantitative studies that investigate how neuromuscular stresses are affected by aging, genetic mutations and drug treatments in this organ and other luminal organs.

18 Oct 2021
Decision trees are important both as interpretable models amenable to
high-stakes decision-making, and as building blocks of ensemble methods such as
random forests and gradient boosting. Their statistical properties, however,
are not well understood. The most cited prior works have focused on deriving
pointwise consistency guarantees for CART in a classical nonparametric
regression setting. We take a different approach, and advocate studying the
generalization performance of decision trees with respect to different
generative regression models. This allows us to elicit their inductive bias,
that is, the assumptions the algorithms make (or do not make) to generalize to
new data, thereby guiding practitioners on when and how to apply these methods.
In this paper, we focus on sparse additive generative models, which have both
low statistical complexity and some nonparametric flexibility. We prove a sharp
squared error generalization lower bound for a large class of decision tree
algorithms fitted to sparse additive models with component functions.
This bound is surprisingly much worse than the minimax rate for estimating such
sparse additive models. The inefficiency is due not to greediness, but to the
loss in power for detecting global structure when we average responses solely
over each leaf, an observation that suggests opportunities to improve
tree-based algorithms, for example, by hierarchical shrinkage. To prove these
bounds, we develop new technical machinery, establishing a novel connection
between decision tree estimation and rate-distortion theory, a sub-field of
information theory.

29 Apr 2020
In the past few years it has been demonstrated that electroencephalography (EEG) can be recorded from inside the ear (in-ear EEG). To open the door to low-profile earpieces as wearable brain-computer interfaces (BCIs), this work presents a practical in-ear EEG device based on multiple dry electrodes, a user-generic design, and a lightweight wireless interface for streaming data and device programming. The earpiece is designed for improved ear canal contact across a wide population of users and is fabricated in a low-cost and scalable manufacturing process based on standard techniques such as vacuum forming,plasma-treatment, and spray coating. A 2.5x2.5 cm^2 wireless recording module is designed to record and stream data wirelessly to a host computer. Performance was evaluated on three human subjects over three months and compared with clinical-grade wet scalp EEG recordings. Recordings of spontaneous and evoked physiological signals, eye-blinks, alpha rhythm, and the auditory steady-state response (ASSR), are presented. This is the first wireless in-ear EEG to our knowledge to incorporate a dry multielectrode, user-generic design. The user-generic ear EEG recorded a mean alpha modulation of 2.17, outperforming the state-of-the-art in dry electrode in-ear EEG systems.
There are no more papers matching your filters at the moment.
